


دستهبندی نشده
آنالیز مرحله اول (Primary Analysis) داده های NGS شامل Filtering و Trimming و معرفی فایل FASTQ
آنالیز اولیه شامل تبدیل داده های خام دستگاه ( سیگنال) به داده های نوکلئوتیدی در قالب توالی های خوانشی می باشد. به عنوان مثال. فایل های FASTQ به طور کلی ، تجزیه و تحلیل اولیه بر روی ابزار NGS انجام می شود ، به عنوان مثال تبدیل پرونده های خام Binary Base Call (BCL) در دستگاه Illumina به داده های توالی بیولوژیکی در قالب میلیون ها خوانش کوتاه.
پردازش سیگنال بین پلت فرم ها متفاوت است ، ابزارهای Illumina سیگنالهای فلورسنت را به توالی نوکلئوتیدی ترجمه می کنند ، در حالی که ابزارهای Ion Torrent سیگنالها را در قالب تغییر pH تبدیل به ولتاژ می کنند. تجزیه و تحلیل اولیه همچنین با اعمال پیش پردازش هایی بر روی خوانش های NGS این اطمینان را می دهد که تنها توالی های مطلوب با طول مناسب و کیفیت بالا وارد مراحل بعدی آنالیز خواهند شد. این مراحل خود دارای سه بخش بوده که به ترتیب شامل:
Filtering:
توالی ها بر اساس کیفیت باز های خوانشی بر پایه امتیاز Phred و طول خوانش فیلتر می شوند چرا که وجود باز ها با کیفیت خوانشی ضعیف با تشخیص یک واریانت به صورت مثبت کاذب همراه بوده همچنین عدم حذف خوانش های بسیار کوتاه اتصال توالی را به نواحی غیر اختصاصی در حین هم ردیف سازی افزایش خواهد داد. امتیاز کیفیت Phred معیاری برای کیفیت شناسایی بازهای نوکلئوتیدی تولید شده توسط توالی یابی DNA خودکار است.
در ابتدا برنامه کامپیوتری Phred برای کمک به اتوماسیون توالی یابی DNA در پروژه ژنوم انسانی توسعه داده شد. امتیازهای کیفیت Phred به هر باز نوکلئوتیدی خوانش شده در ردیابی توالیسنجی خودکار اختصاص داده میشود. فرمت FASTQ نمرات phred را به عنوان کاراکترهای ASCII در کنار دنباله های خوانده شده رمزگذاری می کند.
نمرات کیفیت Phred برای مشخص کردن کیفیت توالی های DNA به طور گسترده پذیرفته شده است و می تواند برای مقایسه کارایی روش های مختلف توالی یابی استفاده شود. در سال 1995، Bonfield و Staden روشی را برای استفاده از امتیازهای کیفی باز ها برای بهبود دقت توالیهای در پروژههای توالییابی DNA پیشنهاد کردند. اولین برنامه ای که امتیازهای کیفی دقیق و قدرتمندی را ایجاد کرد، برنامه Phred بود. Phred قادر به محاسبه نمرات کیفیت بسیار دقیقی بود که به صورت لگاریتمی با احتمالات خطا مرتبط بودند.
Phred به سرعت توسط تمام مراکز اصلی توالی یابی ژنوم و همچنین بسیاری از آزمایشگاه های دیگر پذیرفته شد. اکثریت قریب به اتفاق توالیهای DNA تولید شده در طول پروژه ژنوم انسانی با Phred پردازش شدند.پس از اینکه نمرات کیفیت Phred به استاندارد مورد نیاز در توالی یابی DNA تبدیل شد، سایر سازندگان ابزارهای توالی یابی DNA، از جمله Li-Cor و ABI، معیارهای امتیازدهی کیفیت مشابهی را برای نرم افزار فراخوانی باز خود توسعه دادند.
Demultiplexing:
اولین مرحله تعیین توالی، ساختن یک کتابخانه از DNA یا RNA است. یک کتابخانه حاوی قطعات DNA است که در هر طرف توسط یک آداپتور احاطه شده اند:
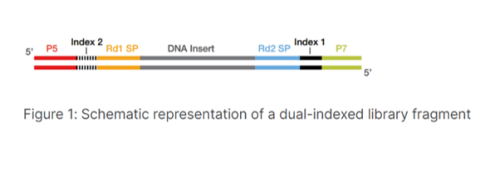
آداپتورها شامل:
- توالی هایی که به کتابخانه اجازه می دهد تا خوشه هایی را در Flow Cell ایجاد کند (توالی های p5 و p7)
- تعیین توالی محل های اتصال پرایمر برای شروع توالی یابی (Rd1 SP و Rd2 SP)
- توالیهای شاخص (شاخص 1 و در صورت لزوم، شاخص 2)، که شناسههای نمونه هستند که امکان ران کردن چند نمونه را در یک Flow Cell فراهم می کنند.
ایلومینا طیف وسیعی از کیتهای آداپتور را ارائه میکند تا انعطافپذیری و استراتژیهای Indexing متعدد را فراهم کند. کیتهای آداپتور Illumina بر اساس تعداد نمونههایی که پشتیبانی میکنند، طول و توالی Index ها، و همچنین شیمی که به وسیله آن به قطعه متصل میشوند، با یکدیگر متفاوت هستند.
در نتیجه زمانی که چند توالی از نمونه های متعدد در یک ران خوانش می شوند با استفاده از بارکدهای مولکولی مختلف به نام Index می بایست از هم تفکیک شوند و در نهایت با استفاده از همین بارکدها از هم قابل افتراق شوند که به این فرآیند Demultiplexing گفته می شود.
Trimming:
دنباله های آداپتور که به انتهای کتابخانه ها در طول فرآیند آماده سازی کتابخانه متصل شده اند ، در این مرحله از خوانش ها حذف می شوند زیرا که وجود آن ها در نقشه برداری و مونتاژ توالی ها تداخل ایجاد می کند. حذف توالی های آداپتور در فرآیندی به نام برش ، یکی از اولین گام ها در تجزیه و تحلیل داده های NGS read trimming یا clipping می باشد. بیش از 30 ابزار برش برای قطع آداپتور وجود دارد که بنابراین انتخاب بهترین ابزار را بسیار راحت می کند.
چرا آداپتورها توالی های ما را آلوده می کنند؟
آداپتورها باید در طول آمادهسازی کتابخانه به تک تک مولکولهای DNA متصل شوند. برای سکانس به روش توالی یابی Illumina، پروتکل های مربوطه (البته در اغلب موارد) شامل یک مرحله قطعه قطعه شدن DNA و به دنبال آن اتصال الیگونوکلئوتیدهای خاص به انتهای 5 و 3 است.
این توالیهای آداپتور 5′ و 3′ دارای توالی های مهمی از جمله توالیهای بارکد، پرایمرهای فوروارد و ریورس(برای توالییابی با انتهای جفت) و توالیهای اتصال مهم برای بیحرکت کردن قطعات روی Flow Cell که امکان تکثیر به وسیله bridge amplification را می دهند می باشند. آلودگی آداپتور منجر به خطاهای alignment NGS و افزایش تعداد خوانش های هم تراز نشده میشود، زیرا توالیهای آداپتور مصنوعی هستند و در توالی ژنومی وجود ندارند.

سلام خواستم بپرسم اگر فلوسل کیت ngsبجای یخچال در فریزر نگهداری شود ممکن است بازم کار کند یا کلا خراب میشود؟
اگر فلوسل بهجای یخچال در فریزر قرار گرفته باشد، احتمال آسیبدیدگی شدید وجود دارد (یخزدگی باعث تخریب کانالها و چیدمان نانوذرات میشود).
📌 نتیجه:
احتمال اینکه کلاً خراب شده باشد زیاد است و استفاده از آن ممکن است منجر به خرابی ران یا داده ناقص شود. توصیه میشود از فلوسل جدید استفاده کنید و با پشتیبانی شرکت سازنده نیز مشورت نمایید.
با سلام و تشکر از مطلب بسیار مفید.