


دستهبندی نشده
تبدیل فایل FASTQ به فایل های SAM و BAM

درباره فایل FASTQ
در طی پروسه هم ردیف سازی تعداد قابل شمارشی از فایل های مختلف ایجاد می شوند که در زیر به ترتیب آورده شده اند:
فایل FASTQ : فایلی با فرمت متنی که شامل توالی های کوتاه خوانش شده به همراه کیفیت هر باز خوانشی است که در مرحله اول به وسیله نرم افزار FASTQC کنترل کیفی انجام شده و نمودارهای حاصل آنالیز خواهند شد. فایل های FASTQ به فرمت استاندارد برای ذخیره داده های NGS از سیستم های توالی یابی Illumina تبدیل شده اند و می توانند به عنوان ورودی برای طیف گسترده ای از مراحل تجزیه و تحلیل داده های ثانویه استفاده شوند.
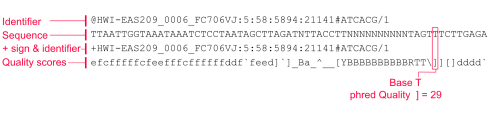
در واقع FASTA به طور اصلی فقط نام یک توالی و خود توالی را ذخیره می کند ولی به طور اختیاری افراد می توانند فیلدهای مد نظر خود را بعد از نام توالی اضافه کنند ولی فرمت FASTQ برای ذخیره توالی و کیفیت مرتبط به آن اختراع شد.
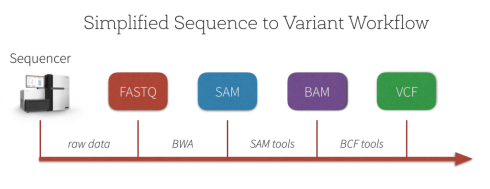
فایل SAM: توالی های موجود در فایل FASTQ با ژنوم مرجع Align شده و اولین فایل خروجی SAM(Sequence Alignment Map) می باشد که مشخص می کند هر کدام از خوانش ها مربوط به کدام بخش از ژنوم می باشند. البته در توالی یابی De novo از هیچ مرجعی برای هم ردیف سازی استفاده نشده و توالی ها براساس شباهت دنباله ها با یک دیگر هم تراز می شوند تا در نهایت یک دنباله طولانی تحت عنوان کانتیگ (Contig) ایجاد شود. در طی پروسه Alignment فایل FASTQ به فایلی با فرمت SAM تبدیل می شود.
فرمت SAM یک فرمت متنی برای ذخیره دادههای سکانس و Align شده به ژنوم مرجع به علاوه دیگر ویژگی ها می باشد. فرمت SAM به عنوان یک نسخه قابل خوانش توسط انسان می باشد که برای استفاده از آن ها در مراحل زیر دست از فرمت BAM که به صورت فشرده و باینری می باشد استفاده می شود. در نتیجه فایلهای SAM و فایلهای BAM حاوی اطلاعات یکسان هستند، اما با فرمت متفاوت.
در حال حاضر، بیشتر دادههای فرمت SAM از aligners خارج میشوند که فایلهای FASTQ را میخوانند و توالیها را به یک موقعیت با توجه به ژنوم مرجع شناخته شده اختصاص میدهند. در آینده، SAM همچنین برای بایگانی دادههای توالی تراز نشده که به صورت مستقیم از ماشینهای توالییابی تولید شده است نیز استفاده می شود.
فایل BAM: فرمت باینری فشرده از فایل SAM می باشد. BAM به طور فزاینده ای به عنوان یک جایگزین برای صرفه جویی در فضا به جای فایل های FASTQ خام استفاده می شود و همه نرم افزارهای Alignment فعلی می توانند SAM/BAM را به عنوان فرمت خروجی تولید کنند. پس از ایجاد فرمت BAM، فایل را می توان ایندکس کرده و دسترسی سریعی به هر منطقه از ژنوم مرجع را فراهم نمود.
هر دو فایل SAM و BAM حاوی یک بخش header اختیاری هستند که به دنبال آن بخش alignments قرار می گیرد.بخش header ممکن است حاوی اطلاعاتی در مورد کل فایل و اطلاعات اضافی برای بخش alignments باشد. بخش alignments حاوی اطلاعات مربوط به هر توالی در مورد اینکه کجا/چگونه با ژنوم مرجع همردیف می شود می باشد.
فایل VCF: فایلی که در آن با مقایسه ژنوم بیمار با ژنوم رفرنس تفاوت ها را ذخیره می کند که در ادامه به وسیله VQSR کنترل کیفی خواهد شد. فرمت Variant Call Format (VCF) فرمت یک فایل متنی مورد استفاده در بیوانفورماتیک را برای ذخیره تغییرات توالی ژن فراهم می کند. این قالب با ظهور پروژه های ژنوتیپ و توالی یابی DNA در مقیاس بزرگ مانند پروژه 1000 ژنوم توسعه یافته است. فرمتهای موجود برای دادههای ژنتیکی مانند (GFF) همه دادههای ژنتیکی را ذخیره میکنند، که بسیاری از آنها زائد هستند زیرا در ژنومها به اشتراک گذاشته میشوند. با استفاده از فرمت VCF، فقط تغییرات باید همراه با ژنوم مرجع ذخیره شوند.
