


بیماری ها
سندرم کاودن

سندرم کاودن چیست؟
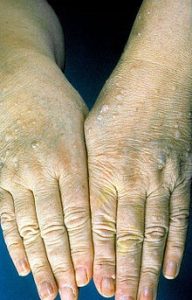
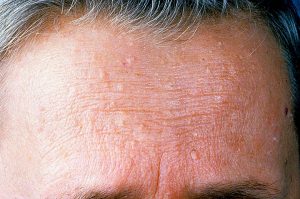
کاودن یک اختلال ژنتیکی است که با تومورهای غیرسرطانی به نام هامارتوم و افزایش خطر ابتلا به برخی سرطانها مشخص میشود.تقریباً همه افراد مبتلا به سندرم کاودن به هامارتوما مبتلا میشوند. این تودهها بیشتر روی پوست و غشاهای مخاطی (مانند مخاط دهان و بینی) دیده میشوند، اما میتوانند در روده و سایر قسمتهای بدن نیز ایجاد شوند. رشد هامارتومها روی پوست و غشاهای مخاطی معمولاً در اواخر دهه بیست سالگی فرد آشکار میشود.
این سندرم با افزایش خطر ابتلا به چندین نوع سرطان، به ویژه سرطان سینه، غدهای در پایین گردن به نام تیروئید، و پوشش داخلی رحم (آندومتر) مرتبط است. سرطانهای دیگری که در افراد مبتلا به سندرم کاودن شناسایی شدهاند عبارتند از: سرطان کلیه، سرطان کولورکتال و یک نوع تهاجمی سرطان پوست به نام ملانوم. در مقایسه با جمعیت عمومی، افراد مبتلا به سندرم کاودن در سنین پایینتر به این سرطانها مبتلا میشوند که اغلب از سی یا چهل سالگی شروع میشود.
افراد مبتلا به این سندرم نیز در مقایسه با جمعیت عمومی بیشتر در معرض ابتلا به بیش از یک سرطان در طول زندگی خود هستند. سایر بیماریهای سینه، تیروئید و آندومتر نیز در این سندرم شایع هستند. علائم و نشانههای اضافی میتواند شامل بزرگ شدن سر (ماکروسفالی) و تومور مغزی نادر و غیر سرطانی به نام بیماری لرمیت-دوکلوس (Lhermitte-Duclos) باشد. درصد کمی از افراد مبتلا دارای تاخیر در رشد، ناتوانی ذهنی یا اختلال طیف اوتیسم هستند که میتواند بر ارتباطات و تعامل اجتماعی تأثیر بگذارد.
برخی از افراد معیارهای دقیق برای تشخیص بالینی این سندروم را ندارند، اما برخی از ویژگیهای مشخصه این بیماری، به ویژه سرطانها را دارند. این افراد اغلب به عنوان مبتلا به سندرم شبه کاودن توصیف میشوند. هر دو سندرم کاودن و سندرم شبه کاودن به دلیل جهش در ژن های مشابه ایجاد میشوند.
ویژگیهای این سندرم با اختلال دیگری به نام سندرم Bannayan-Riley-Ruvalcaba همپوشانی دارد. افراد مبتلا به سندرم Bannayan-Riley-Ruvalcaba نیز دچار هامارتوما و سایر تومورهای غیرسرطانی میشوند. برخی از افراد مبتلا به این سندرم دارای بستگانی هستند که به سندرم Bannayan-Riley-Ruvalcaba مبتلا هستند و سایر افراد مبتلا ویژگیهای مشخصه هر دو بیماری را دارند. بر اساس این شباهتها، پژوهشگران پیشنهاد کردهاند که نشانگان کاودن و سندرم Bannayan-Riley-Ruvalcaba طیفی از ویژگیهای همپوشانی شناخته شده به عنوان سندرم تومور هامارتوم PTEN نشان میدهند.
فراوانی این اختلال
اگرچه شیوع دقیق این بیماری ناشناخته است، محققان تخمین میزنند که از هر 200 هزار نفر، 1 نفر را تحت تاثیر قرار میدهد.
علت
تغییرات در ژن PTEN، KLLN یا WWP1 معمولاً در افراد مبتلا به سندرم کاودن یا سندرم شبه کاودن شناسایی میشود.
حدود 25 درصد از این بیماری و درصد کمی از موارد سندرم شبه کاودن ناشی از جهش در ژن PTEN است. پروتئین تولید شده از ژن PTEN یک سرکوب کننده تومور است، به این معنی که به طور معمول از رشد و تقسیم (تکثیر) خیلی سریع یا به روش کنترل نشده سلولها جلوگیری میکند. جهش در ژن PTEN از تنظیم موثر پروتئین PTEN جلوگیری میکند که منجر به تقسیم سلولی کنترل نشده و تشکیل هامارتوم ها و تومورهای سرطانی میشود. ژن PTEN احتمالاً عملکردهای مهم دیگری در داخل سلول دارد. با این حال، برای تعیین نقش جهش در این ژن در ایجاد سایر ویژگیهای سندرم کاودن، مانند ماکروسفالی و ناتوانی ذهنی، تحقیقات لازم است.
به ندرت، سندرم کاودن و سندرم شبه کاودن ناشی از تغییر ژن KLLN است. این ژن دستورالعملهایی را برای ساخت پروتئینی به نام کیلین (Killin) ارائه میدهد. مانند پروتئین تولید شده از ژن PTEN، کیلین احتمالا به عنوان یک سرکوب کننده تومور عمل میکند. تغییر ژنتیکی که باعث سندرم کاودن و سندرم شبه کاودن شده، منجر به کاهش تولید پروتئین کیلین میشود. کاهش مقدار کیلین به سلولهای غیرطبیعی اجازه زنده ماندن و تکثیر نامناسب را داده که منجر به تشکیل تومور میشود.
درصد کمی از سندرم کاودن و سندرم شبه کاودن با انواع ژن WWP1 مرتبط است. ژن WWP1 دستورالعملهایی برای ساخت پروتئینی ارائه داده که در فرآیندی که پروتئینهای دیگر را هدف قرار میدهد تا در سلولها تجزیه شوند، دخیل است.
در طی این فرآیند، پروتئین WWP1 به پروتئین PTEN متصل میشود که عملکرد PTEN را مختل میکند. انواع ژن WWP1 به عنوان “به دست آوردن عملکرد” توصیف میشوند زیرا به نظر میرسد فعالیت پروتئین WWP1 را افزایش میدهند. مطالعات نشان میدهند که پروتئین تغییریافته راحتتر از حالت عادی به پروتئین PTEN متصل میشود. اتصال بیش از حد، فعالیت سرکوبگر تومور PTEN را مختل کرده و به سلولها اجازه میدهد بدون کنترل تکثیر شوند و منجر به تشکیل تومور شود.
جهش در چند ژن دیگر هر کدام مسئول درصد بسیار کمی از موارد سندرم کاودن و سندرم شبه کاودن هستند. در بقیه موارد، علت ژنتیکی ناشناخته است.
الگوی وراثت
این بیماری و سندرم شبه کاودن در یک الگوی اتوزومال غالب به ارث می رسند، به این معنی که یک کپی از ژن تغییر یافته در هر سلول برای ایجاد این بیماری و افزایش خطر ابتلا به سرطان کافی است. در برخی موارد، یک فرد مبتلا جهش را از یکی از والدین مبتلا به ارث میبرد. موارد دیگر ممکن است ناشی از جهشهای جدید در ژن باشد. این موارد در افرادی رخ میدهد که سابقه این اختلال را در خانواده خود ندارند.
نامهای دیگر این اختلال
- CD
- بیماری کاودن
- CS
- MHAM
- سندرم هامارتوم مالتیپل
خلاصه:
Cowden syndrome اختلالی است که با ایجاد تعداد زیادی زوائد تومور مانند غیرسرطانی به نام هامارتوم (hamartoma) و افزایش خطر ابتلا به سرطانهای خاصی، مشخص میشود. تقریباً تمام مبتلایان به سندروم کاودن، دچار هامارتوم میشوند. این زوائد اغلب بر روی پوست و غشا مخاطی (mucous membranes) (مثل پوشش دهان و بینی) ایجاد میشوند، اما ممکن است در سایر بخشهای بدن مانند روده (the intestine) نیز ایجاد شوند.
مترجم: امید آهنگریان ابهری
مطالعه صدها مطلب علمی در حوزه بیولوژی
آرشیو جدیدترین خبرهای روز دنیای بیولوژی
