


بیماری ها
سندرم Aicardi-Goutieres (آیکاردی گوتیرز)
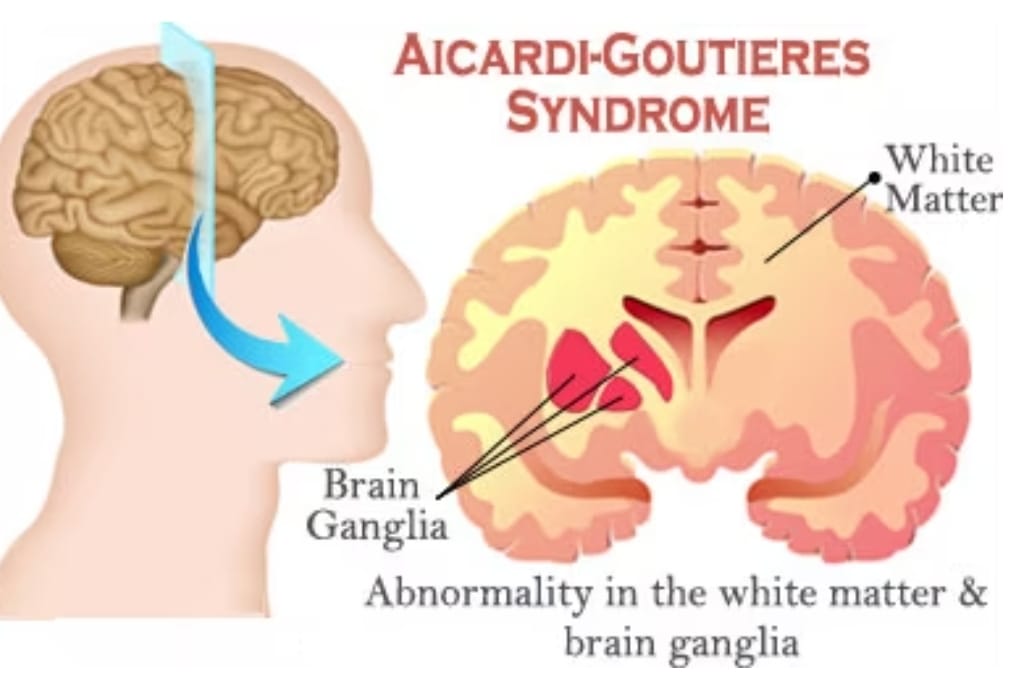
شرح بیماری :سندرم Aicardi-Goutieres (AGS) (آیکاردی گوتیرز) یک آنسفالوپاتی (آسیب مغزی) ارثی است که بر نوزادان تازه متولدشده تأثیر میگذارد و معمولاً منجر به نارسایی شدید ذهنی و جسمی میشود. دو حالت از این سندرم وجود دارد: یک فرم زودرس که شدید است و یک فرم دیررس که تأثیر کمتری بر عملکرد عصبی دارد. فرم زودرس بر حدود 20 درصد از تمام نوزادان مبتلا به AGS اثر میکند. این نوزادان با ناهنجاریهای عصبی و کبدی، مانند بزرگشدگی کبد و طحال و افزایش بیش از حد آنزیمهای کبدی متولد میشوند. رفتار وحشتزده و میل کم آنها به تغذیه، شبیه (به مثابهی تقلیدی از) حالت عفونت ویروسی مادرزادی است. به عبارتی دیگر، علیرغم این که مجموع این علائم و نشانهها، معمولاً با واکنش سیستم ایمنی بدن نسبت به عفونت ویروسی که در بدو تولد وجود دارد (مادرزادی) مرتبط اند، هیچ عفونت واقعی در این نوزادان یافت نمیشود. به همین دلیل، گاهی اوقات از سندرم Aicardi-Goutières به عنوان “تقلید عفونت مادرزادی” یاد میشود.
نوزادان مبتلا به AGS دیررس، پس از هفتهها یا ماههای آغازینی که رشدونمو طبیعی داشتهاند، به مرور علائمی را بروز میدهند که به صورت کاهش تدریجی رشد سر، ضعیف یا منقبض شدن عضلات (اسپاسم عضلانی)، و عقبماندگیهای شناختی و رشدی متوسط تا شدید، پدیدار میشوند.
در طی سال اول زندگی، اکثر افراد مبتلا به سندرم Aicardi-Goutières یک دوره اختلال شدید مغزی (آنسفالوپاتی) را تجربه میکنند که معمولاً چندین ماه طول میکشد. در طول مرحله آنسفالوپاتیک این بیماری، نوزادان مبتلا معمولاً به شدت تندخو هستند و به خوبی غذا نمیخورند. آنها ممکن است ضمن این که عفونتی در کار نیست، دچار تبهای نامنظم شوند (sterile pyrexias) و تشنّج داشتهباشند. آنها دیگر مهارتهای جدیدی کسب نمیکنند و رفتهرفته تواناییهایی را که قبلاً به دست آورده بودند، از دست میدهند(پسرفت رشد). همچنین روند
رشد مغز و جمجمه کاهش مییابد که منجر به کوچک شدن غیرطبیعی اندازه سر میشود (میکروسفالی). در این مرحله از بیماری، گلبولهای سفید و سایر مولکولهای مرتبط با التهاب سیستم ایمنی را میتوان در مایع مغزی نخاعی، مایعی که مغز و نخاع (سیستم عصبی مرکزی) را احاطه کردهاست، مشاهده کرد. این یافتههای غیرطبیعی مرتبط با التهاب و آسیب بافتی در سیستم عصبی مرکزی است.
مرحله آنسفالوپاتیک سندرم Aicardi-Goutieres (آیکاردی گوتیرز) باعث آسیب عصبی دائمی میشود که معمولاً شدید است. تصویربرداری پزشکی فقدان ماده سفید در مغز را نشان میدهد (لوکودیستروفی). ماده سفید متشکل از رشتههای عصبیست که توسط میلین- ماده ای که از اعصاب محافظت کرده و موجب انتقال سریع تکانههای عصبی میشود- پوشانده شدهاست. همچنین افراد مبتلا دارای رسوب غیرطبیعی کلسیم (کلسیفیکاسیون) در مغز هستند. در نتیجهی این آسیب عصبی، اکثر افراد مبتلا به سندرم Aicardi-Goutières دارای ناتوانی ذهنی جدّی هستند. آنها همچنین دارای انقباض عضلانی (حالت اسپاسمی)، کشش غیرارادی عضلات مختلف (دیستونی یا اختلال در تونوس عضلانی) به ویژه در عضلات بازو و تونوس عضلانی پایین (هیپوتونی یا کاهش کشیدگی عضلات) در بالاتنه میباشند.
برخی از افراد مبتلا به سندرم Aicardi-Goutieres (آیکاردی گوتیرز) دارای ویژگیهای بارز اختلالات خودایمنی هستند که زمانی رخ میدهند که سیستم ایمنی بدن دچار مشکل شده و به سیستمها و اندامهای خودی بدن حمله میکند. برخی از این ویژگیها با اختلال دیگری به نام لوپوس اریتماتوز سیستمیک (SLE) همپوشانی دارند. یکی از ویژگی های SLE که در حدود 40 درصد از افراد مبتلا به سندرم Aicardi-Goutières نیز ظاهر میشود، یک مشکل پوستی به نام Chilblain (سرمازدگی) است. Chilblains ضایعات پوستی دردناک و خارشدار هستند که متورّم و قرمزند و معمولاً روی انگشتان دست و پا و گوش ظاهر میشوند. آنها در پی التهاب رگهای خونی کوچک شکل گرفته و احتمالاً در اثر قرار گرفتن در معرض سرما ایجاد شده یا بدتر میشوند. مشکلات بینایی، سفتی مفاصل و زخمهای دهان از دیگر ویژگیهایی هستند که میتوانند در هر دوی این بیماریها ایجاد شوند.
در نتیجهی مشکلات عصبی شدید که معمولاً با سندرم Aicardi-Goutières همراه است، اکثر افراد مبتلا به این بیماری بیشتر از دوران کودکی زنده نمیمانند. با این وجود، برخی از افراد مبتلا که دیرتر به این بیماری دچار میشوند یا مشکلات عصبی خفیفتری دارند، تا بزرگسالی زندگی میکنند.
علت بیماری
جهش در چهار ژن مختلف با AGS مرتبط است:
AGS1 و AGS5 (فرم اتوزوم مغلوب) ناشی از جهش در ژن TREX1 هستند.
AGS2 ناشی از جهش در ژن RNASEH2B است.
AGS3 ناشی از جهش در ژن RNASEH2C است.
و AGS4 به دلیل جهش در ژن RNASEH2A ایجاد میشود.
ژنهای TREX1، RNASEH2A، RNASEH2B و RNASEH2C دستورالعملهایی را برای ساختن نوکلئازها، ارائه میدهند. نوکلئازها آنزیمهایی هستند که به تجزیه مولکولهای DNA و ایزومر آن RNA زمانی که دیگر مورد نیاز نیستند، کمک میکنند. این مولکولها یا قطعات DNA و RNA ممکن است در طی مرحله اول تولید پروتئین (رونویسی)، تکثیرِ (همانندسازی) مواد ژنتیکی سلولها برای آمادهسازی تقسیم سلولی، ترمیم DNA، مرگ سلولی (آپوپتوز) و سایر فرآیندها تولید شوند. اعتقاد بر این است که جهش در هر یک از این ژنها منجر به فقدان یا عملکرد غیرطبیعی آنزیم نوکلئاز مربوطه میشود. محققان اظهار دارند که فقدان یا اختلال در عملکرد آنزیم ممکن است منجر به تجمّع DNA و RNA غیرضروری در سلولها شود. سلولها ممکن است DNA و RNA غیرضروری را با مواد ژنتیکی مهاجمان ویروسی اشتباه گرفته و باعث واکنشهای سیستم ایمنی در قسمتهای مختلف بدن شود که منجر به آنسفالوپاتی، آسیب پوستی و سایر علائم و نشانههای سندرم Aicardi-Goutières میشود.
جهشها در ژنهایی دیگر از جمله SAMHD1، IFIH1 و ADAR نیز میتوانند موجب سندرم Aicardi-Goutieres (آیکاردی گوتیرز)شوند. این ژنها دستورالعملهایی را برای ساخت پروتئینهایی ارائه میدهند که در سیستم ایمنی دخیلاند. جهش در این ژنها باعث فعالسازی نامناسب پاسخ ایمنی بدن و در نتیجه آسیب التهابی به مغز، پوست و سایر سیستمهای بدن شده که منجر به ویژگیهای بارز سندرم Aicardi-Goutières میگردد.
الگوی وراثت
سندرم Aicardi-Goutières میتواند الگوهای وراثتی گوناگونی داشته باشد. در بیشتر مواردِ ناشی از جهش در ژنهای ADAR، TREX1، RNASEH2A، RNASEH2B، RNASEH2C و SAMHD1 بیماری در الگوی اتوزومی مغلوب به ارث میرسد، به این معنی که هر دو نسخه از ژن در هر سلول، جهشیافته است. والدین یک فرد مبتلا به بیماری اتوزومی مغلوب هر کدام یک نسخه از ژن جهشیافته را دارند، اما معمولاً علائم و نشانههای این بیماری را بروز نمیدهند.
هنگامی که سندرم Aicardi-Goutières به دلیل جهش در ژن IFIH1 یا جهشهای شدید در ژن TREX1 یا ADAR ایجاد شود، طبق الگوی اتوزومی غالب به ارث میرسد، به این معنی که یک نسخه از ژن تغییریافته در هر سلول برای ایجاد بیماری کافی است. این موارد ناشی از جهشهایی جدید در ژن است و در افرادی رخ میدهد که سابقه این بیماری را در خانوادهشان ندارند.
مترجم: ستایش بصیری
