


اطلاعات عمومی
RNA-seq در بیوانفورماتیک
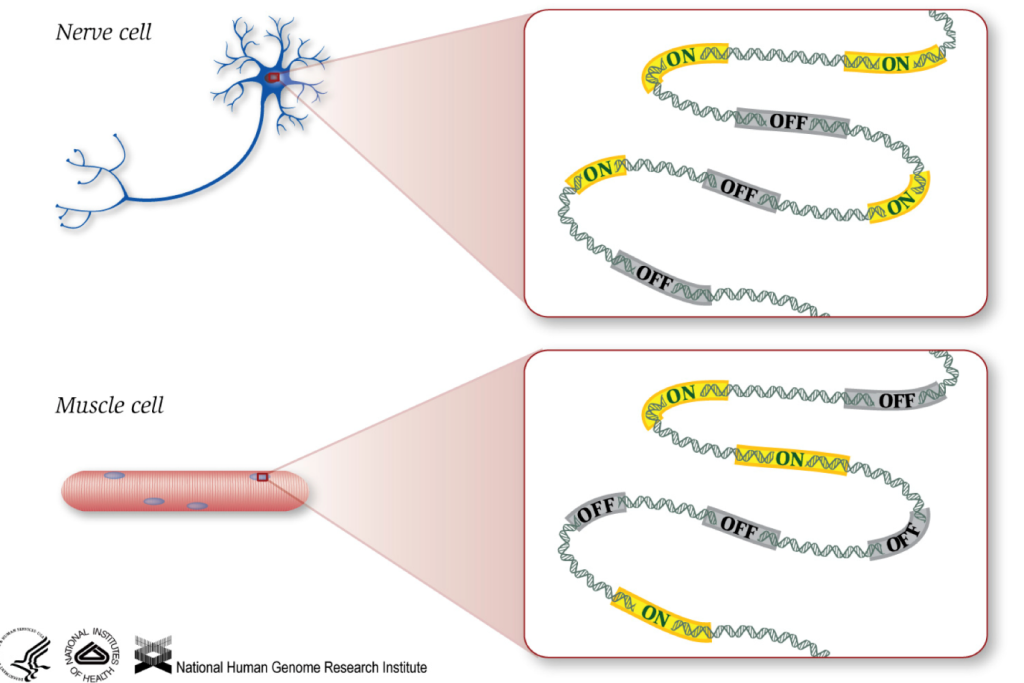
یکی از روش های مطالعه ی ترانسکریپتوم که براساس تکنولوژی های نسل جدید توالی یابی انجام می شود، روش RNA-seq می باشد. RNA-seq یک تکنیک تجربی مهیج است که برای کاوش و یا تعیین کمیت بیان ژن تحت شرایط مختلف مورد استفاده قرار می گیرد. همانطور که می دانیم ، ژن ها دستورالعمل هایی برای ساخت پروتئین ها ارائه می دهند، پروتئین هایی که عملکردی در سلول دارند. اگرچه همه سلولها دارای توالی DNA یکسانی هستند ، اما سلولهای عضلانی به دلیل ژنهای مختلفی که در این سلولها روشن شده و RNA ها و پروتئینهای مختلف تولید شده از سلولهای عصبی و انواع دیگر سلولها متفاوت است.
فرایندهای مختلف بیولوژیکی و همچنین جهش ها می توانند بر روی ژن هایی که روشن و خاموش هستند تأثیر بگذارند ، علاوه بر این می توانند تعیین کنند که ژن های خاص چه مقدار خاموش یا روشن هستند.
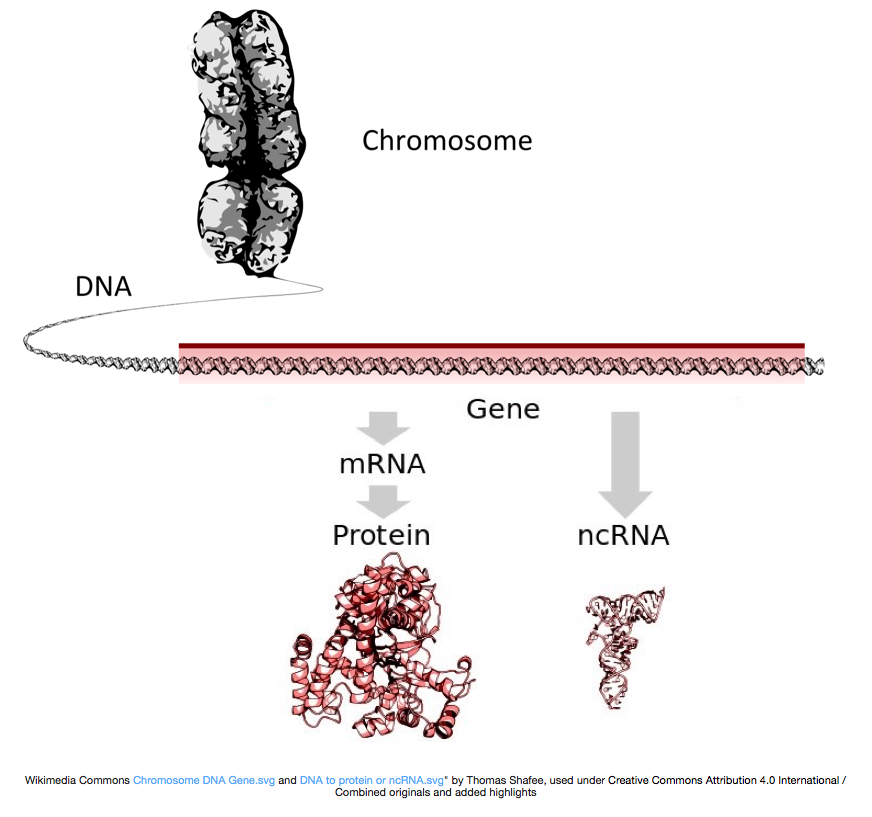
برای ساخت پروتئین ، DNA به RNA پیام رسان یا mRNA رونویسی می شود که توسط ریبوزوم به پروتئین تبدیل می شود. با این حال ، برخی از ژن ها RNA را رمزگذاری می کنند که به پروتئین تبدیل نمی شود. به این RNA ها RNA غیر کد کننده یا ncRNA گفته می شود. غالباً این RNA ها عملکردی به خودی خود دارند و شامل rRNA ها ، tRNA ها و siRNA ها می شوند. تمام RNA های رونویسی شده از ژن ها، رونوشت نامیده می شوند.
RNA برای تبدیل به پروتئین، باید تحت پردازش قرار گیرد. در شکل زیر ، رشته بالا در تصویر نشان دهنده یک ژن در DNA است که از مناطق ترجمه نشده (UTR) و قالب خوانش باز تشکیل شده است. ژن ها به pre-mRNA رونویسی می شوند که هنوز حاوی توالی های اینترونی هستند. پردازش های پس از رونویسی شامل اضافه شدن یک دم پلی A، کلاهک 5’ و حذف اینترون ها می باشد که منجر به تولید mRNA بالغ گردیده که در نهایت می تواند به پروتئین تبدیل شود.
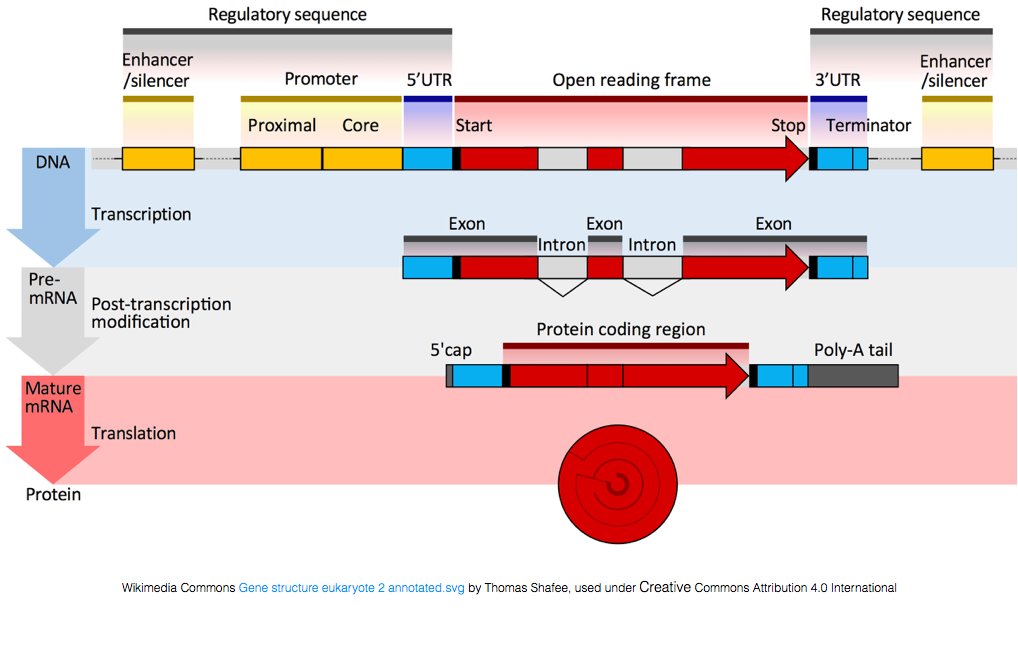
در حالی که رونوشت های mRNA دارای دنباله polyA هستند ، بسیاری از رونوشت های RNA غیر کد کننده این کار را نمی کنند زیرا پردازش پس از رونویسی برای این نسخه ها متفاوت است.
ترانسکریپتومیکس
ترانسکریپتوم به عنوان مجموعه ای از همه رونوشت های موجود در سلول تعریف می شود. از داده های RNA-seq می توان برای کشف و یا تعیین کمیت رونویسی از موجودات زنده استفاده کرد که می تواند برای انواع آزمایش های زیر مورد استفاده قرار گیرد:
بیان ژن افتراقی: ارزیابی کمی و مقایسه سطوح بیانی
مونتاژ رونویسی یا assembly: ساختن مشخصات مناطق رونویسی شده ژنوم ، ارزیابی کیفی.
می تواند با استفاده از assembly به ساخت مدل های ژنی بهتر و تأیید آنها کمک نماید.
Metatranscriptomics یا تجزیه و تحلیل رونوشت ها.
تهیه کتابخانه Illumina
هنگام شروع آزمایش RNA-seq ، برای هر نمونه RNA باید استخراج و به منظور تعیین توالی تبدیل به کتابخانه cDNA شود. گردش کار کلی برای تهیه کتابخانه در تصاویر گام به گام زیر شرح داده شده است:
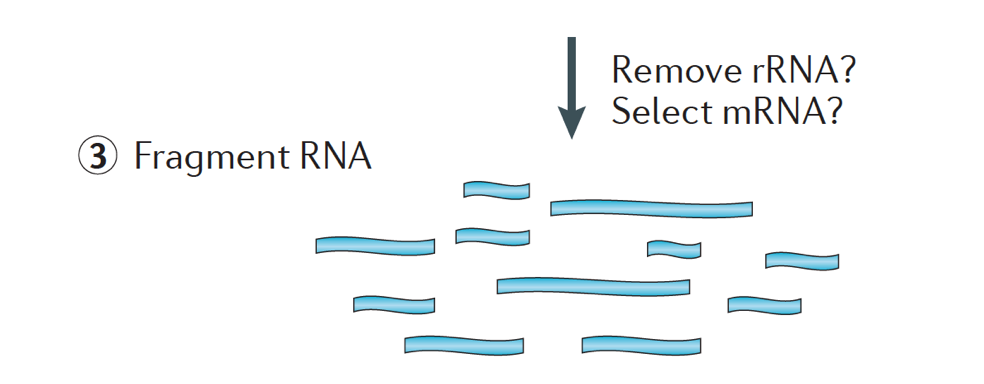
به طور خلاصه ، RNA از نمونه استخراج شده و آلودگی DNA با DNase حذف می شود.
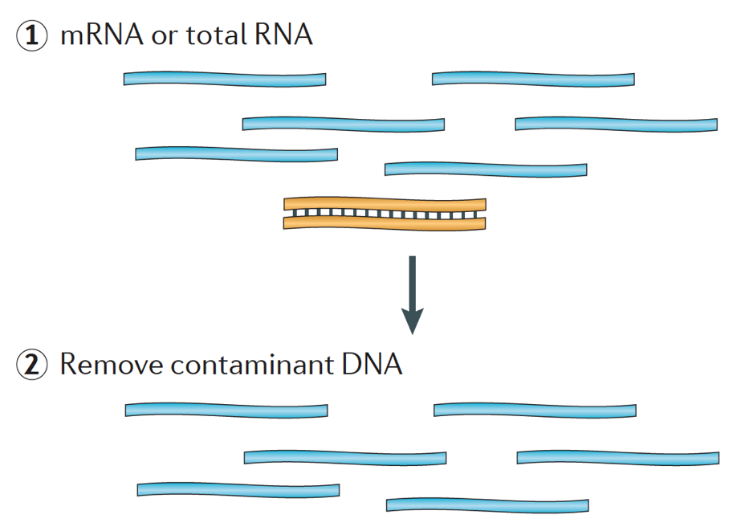
نمونه های RNA بعدا تحت انتخاب mRNA و یا حذف rRNA قرار می گیرند. در نهایت RNA های حاصل قطعه قطعه می شوند.
به طور کلی ، RNA ریبوزومی نشان دهنده اکثر RNA های موجود در سلول است ، در حالی که RNA های پیام رسان یا mRNA درصد کمی از RNA کل (تقریبا 2 درصد) را در انسان نشان می دهند. بنابراین ، اگر می خواهیم ژن های کد کننده ی پروتئین را مطالعه کنیم ، باید mRNA را غنی کنیم یا rRNA را تخلیه کنیم. برای تجزیه و تحلیل بیان افتراقی ژن، بهتر است mRNA غنی شود، مگر اینکه قصد داشته باشید اطلاعاتی در مورد RNA های غیر کدکننده یا lnc-RNA ها بدست آورید، پس یک تخریب RNA ریبوزومی انجام دهید.
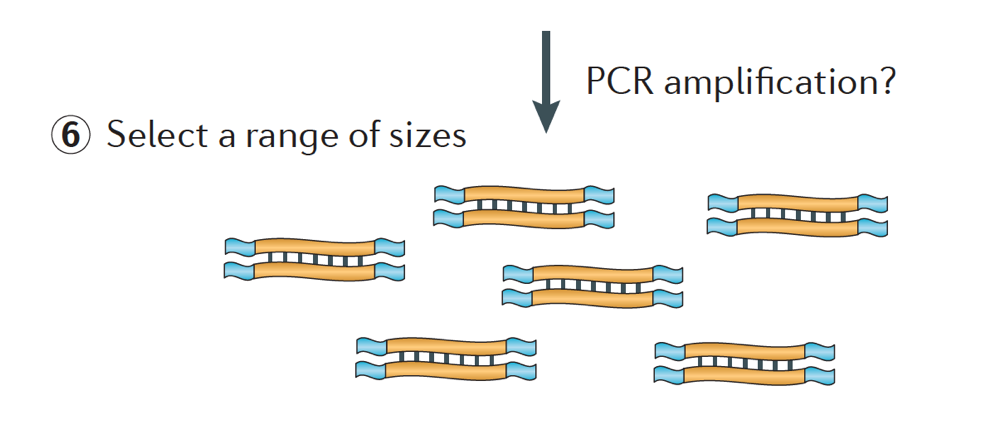
اندازه قطعات هدف در کتابخانه نهایی یک پارامتر اصلی برای ساخت کتابخانه است. قطعه قطعه شدن DNA به طور معمول با روش های فیزیکی (به عنوان مثال ، برش صوتی و فراصوتی) یا روش های آنزیمی (به عنوان مثال ، کوکتل های اندونوکلئاز غیر اختصاصی و واکنش های افزودن برچسب ترانسپوزاز) انجام می شود.
سپس RNA به cDNA دو رشته ای رونویسی می شود و آداپتورهای توالی به انتهای قطعات اضافه می شوند. کتابخانه های cDNA می تواند به گونه ای تولید شوند که بتواند اطلاعات مربوط به اینکه RNA از کدام رشته DNA رونویسی شده است را حفظ کنند. به كتابخانه هايي كه اين اطلاعات را نگهداري مي كنند ، كتابخانه هاي رشته اي یا stranded libraries مي گويند كه اكنون با كيت هاي RNA-Seq رشته اي Illumina’s TruSeq استاندارد هستند. کتابخانه های رشته ای نباید گرانتر از کتابخانه های غیر رشته ای باشند، بنابراین در واقع هیچ دلیلی برای عدم دستیابی به این اطلاعات اضافی وجود ندارد.
3 نوع کتابخانه cDNA موجود است:
Forward (رشته دوم) – خوانش ها یا reads شبیه توالی ژن یا رشته ی دوم cDNA هستند.
Reverse (رشته اول) – خوانش ها یا reads شبیه مکمل توالی ژن و یا رشته ی اول cDNA (TruSeq) هستند.
غیر رشته ای یا Unstranded
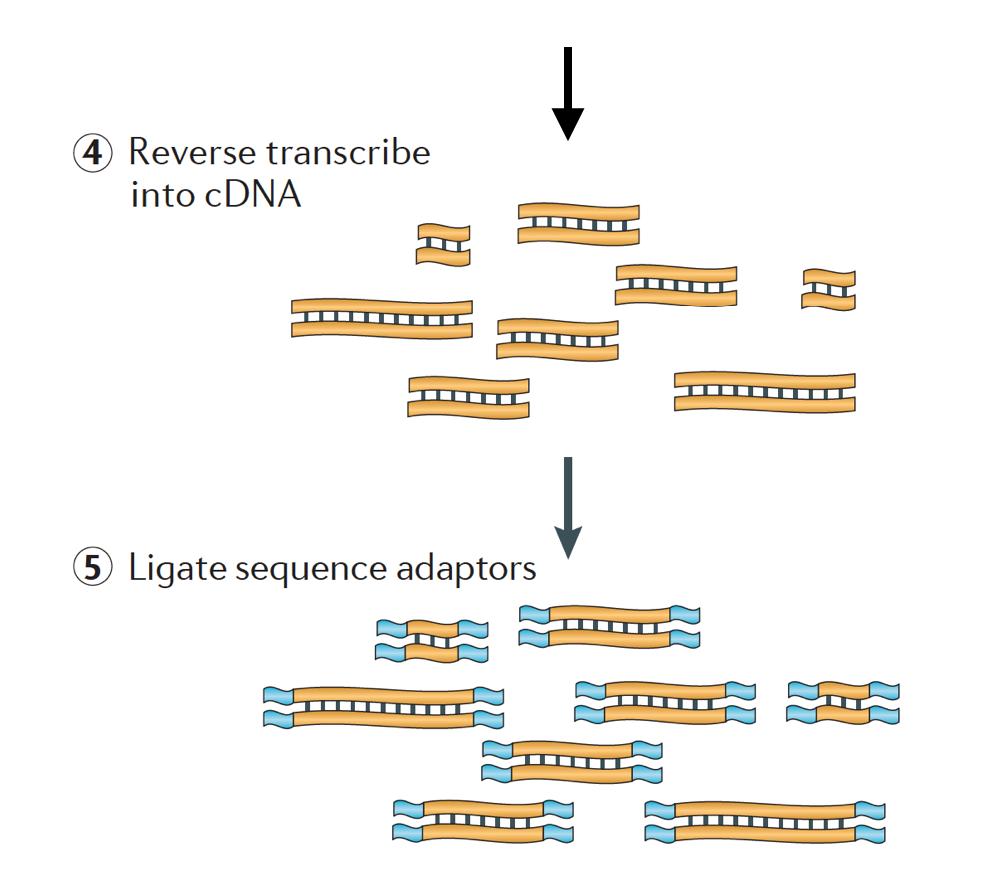
سرانجام ، قطعات در صورت لزوم با PCR تکثیر می شوند ، و قطعات براساس اندازه برای کتابخانه انتخاب می شوند (معمولا 500-300 جفت باز).
توالی یابی Illumina
خوانش های single-end در مقابل خوانش های Paired-end
پس از آماده سازی کتابخانه ها ، می توان توالی یابی را برای تولید توالی های نوکلئوتیدی انتهای قطعات انجام داد که به ان ها خوانش یا reads گفته می شود. شما می توانید یک انتهای قطعات cDNA (single-end reads) یا هر دو انتهای قطعات (Paired-end reads) را توالی یابی نمایید.

به طور کلی توالی یابی single-end کافی است مگر اینکه خوانش ها با مکانهای مختلف ژنوم مطابقت داشته باشند (به عنوان مثال ارگانیسم هایی که دارای ژنهای پارالوگ زیادی هستند) توجه داشته باشید که خوانش های Paired-end به طور کلی 2 برابر گران تر هستند.
پلتفرم های مختلف توالی یابی
انواع پلفرم های Illumina برای انتخاب توالی کتابخانه های cDNA وجود دارد.

تفاوت در پلتفرم می تواند طول خوانش های ایجاد شده، کیفیت خوانش ها، و همچنین تعداد کل خوانش های توالی یابی شده در هر run دستگاه و مقدار زمان لازم برای توالی یابی کتابخانه ها را تغییر دهد. پلتفرم های های مختلف هر یک از یک flow cell متفاوت استفاده می کنند ، که یک سطح شیشه ای است که با آرایش الیگوهای زوج یا Paired oligos پوشانده شده و مکمل آداپتورهای اضافه شده به رشته ی الگو هستند. Flow cell مکانی است که واکنش توالی یابی در آن اتفاق می افتد.

تعیین توالی توسط سنتز
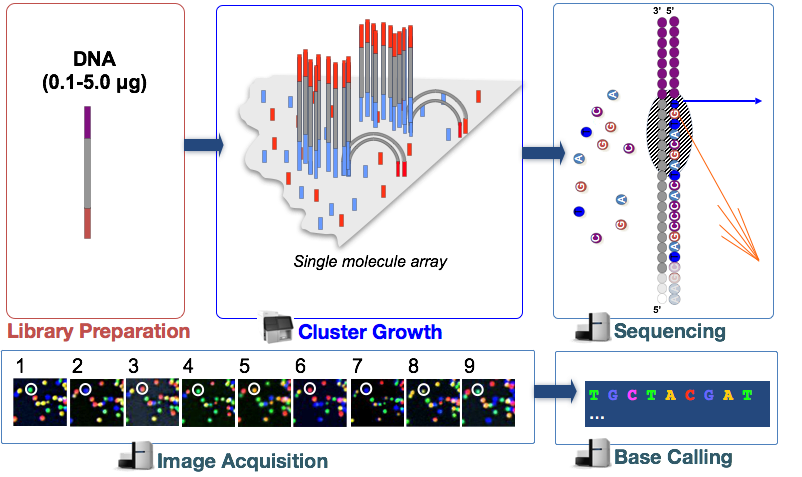
فناوری تعیین توالی Illumina از رویکرد تعیین توالی توسط سنتز استفاده می کند که در زیر با جزئیات بیشتری شرح داده شده است.
در مرحله اول ، قطعات DNA در کتابخانه cDNA دناتوره یا تک رشته ای شده و بر flow cell شیشه ای اعمال می شود. این قطعات دناتوره شده به الیگوهای مکمل متصل می شوند که قبلا به صورت کووالانسی به flow cell متصل شده اند.
ایجاد cluster
پس از اتصال قطعات، فازی به نام ایجاد cluster آغاز می شود. در طی این مرحله، قطعات منفرد به صورت کلنی تکثیر می شوند تا یک cluster از قطعات یکسان ( قطعات نزدیک بهم)ایجاد شود. مرحله ی ایجاد cluster یک مرحله ی ضروری می باشد چرا که فلورسانس می تواند به جای یک قطعه واحد، از یک cluster گرفته شود.
سنتز مکمل با پلیمراز
dsDNA دناتوره می شود و DNA اصلی شسته می شود و رشته سنتز شده به صورت کووالان به flow cell متصل باقی می ماند. تک رشته ای سنتز شده با آداپتورهای مجاور یک پل تشکیل می دهند و dsDNA توسط پلمراز گسترش می یابد. هر رشته به صورت کوالانسی به آدابتور مختلف متصل می شود. این مراحل چندین بار تکرار می شوند تا تمام قطعات منحصر به فرد روی flow cell به صورت کلونی افزایش یابند و cluster هایی از توالی یکسان تشکیل شوند.
تعیین توالی توسط سنتز (و دستیابی به تصویر)
پس از ایجاد cluster، نوکلئوتیدهای دارای برچسب فلورسنت یک به یک گنجانده می شوند (از نظر سیکلی) و تصاویر فلورسانس به منظور شناسایی اینکه کدام نوکلئوتید در هر cluster و در هر سیکل ثبت می شود، گرفته می شود.
Cluster ها دناتوره شده و انتهای 3 پریم برای جلوگیری از سنتزهای نخواسته، بلوکه می شود.
پرایمرهای توالی یابی براساس توالی آداپتورها، اضافه می گردد.
چهار NTP با مارکرهای فلورسنت و توالی های خاتمه دهنده و پلیمراز افزوده می شوند.
پس از تلفیق NTP، cluster توسط یک منبع نوری تحریک می شود و یک سیگنال فلورسنت مشخص، صاتع می شود.
رنگ ثبت می شود، سپس ترمیناتور روی رنگ شکسته و شسته می شود. فرآیند برای تعداد مشخصی چرخه تکرار می شود.
شمارش باز ها یا base calling
Illumina دارای یک نرم افزار اختصاصی است که تمام تصاویر گرفته شده در مرحله قبل را مرور می کند و فایلهای متنی حاوی اطلاعات هر cluster براساس فلورسنت را ایجاد می کند. این نرم افزار علاوه بر شمارش باز ها، امتیازی می دهد که مشخص می کند با چه اطمینانی بازهای A، T، G و C خوانش می شوند.
در صورت وجود ابهامات ، به عنوان مثال در یک چرخه خاص ، تصویر یک cluster دارای رنگ مشخصی نیست که بتواند با یک نوکلئوتید خاص مرتبط باشد ، نرم افزار شمارش بازها “N” را به جای نوکلئوتیدهای A، T، C و یا G قرار می دهد.
تعداد خوشه ~ = تعداد خوانش ها یا reads
تعداد چرخه های توالی = طول خوانش ها یا reads
تعداد چرخه ها (طول خوانش ها) به پلتفرم توالی یابی مورد استفاده و همچنین تنظیمات انتخابی شما بستگی دارد.
Multiplexing
بسته به پلتفرم Illumina (MiSeq, HiSeq, NextSeq)، تعداد لاین ها در هر flow cell و تعداد خوانش ها یا reads دریافتی از هر لاین، بسیار متفاوت می باشد. شما باید تعداد خوانش ها یا reads هر نمونه را انتخاب کنید (به عنوان مثال عمق توالی یابی) و سپس براساس پلتفرمی که انتخاب می کنید، می توانید تعداد کل لاین های مورد نیاز برای مجموعه نمونه های خود را محاسبه کنید.
به طور معمول ، هزینه های تعیین توالی برای هر لاین از flow cell می باشد، شما قادر خواهید بود چندین نمونه را در هر لاین اجرا کنید. بنابراین Illumina روش خوبی برای multiplexing ابداع کرده است که به شما امکان می دهد کتابخانه های چندین نمونه را به طور همزمان در یک لاین flow cell توالی یابی نمایید.
این روش نیاز به افزودن شاخص ها (درون آداپتور Illumina) یا بارکد ویژه (خارج از آداپتور Illumina) دارد که در شماتیک زیر توضیح داده شده است.
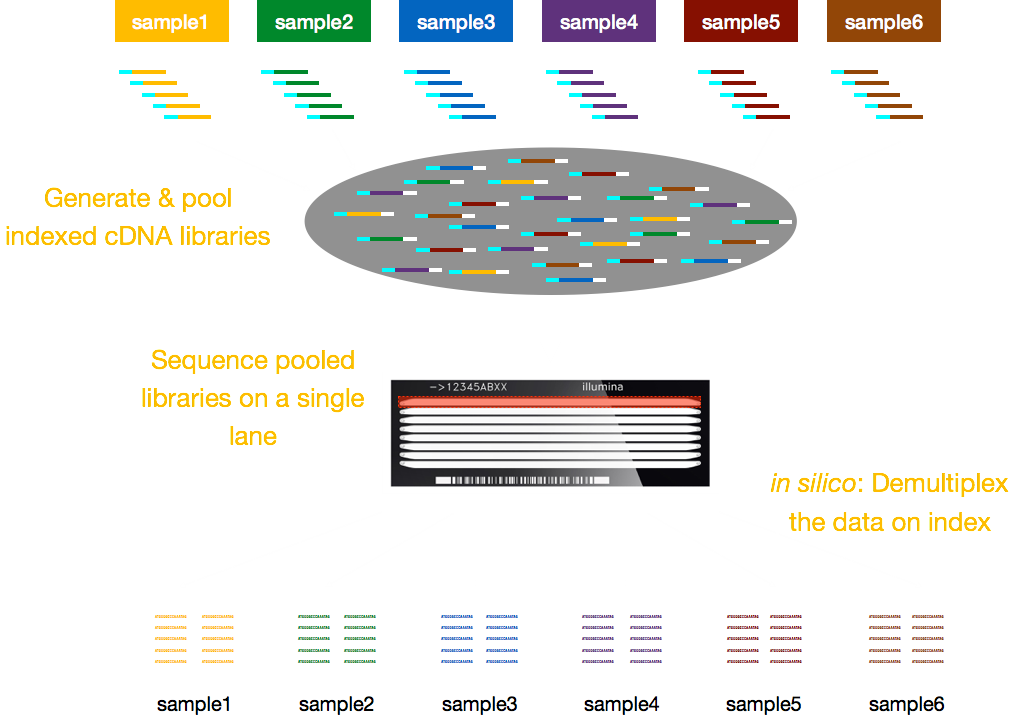
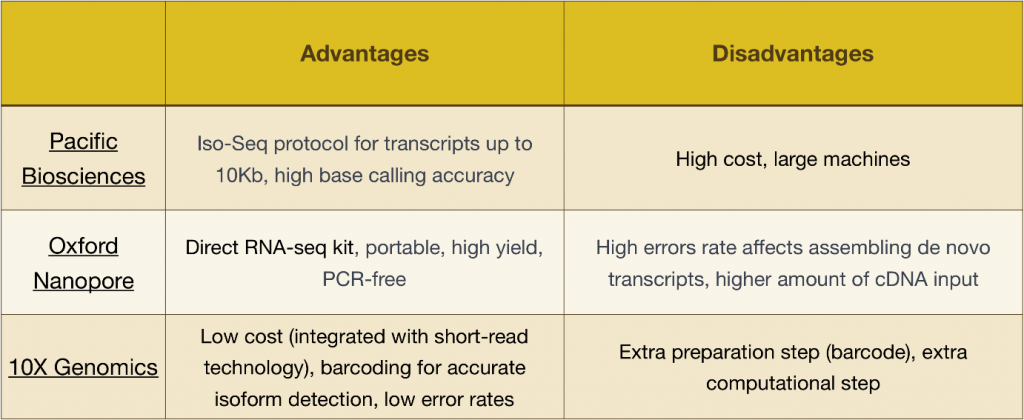
توجه داشته باشید که مراحل ارائه شده در این مبحث مختص به توالی Illumina است که در حال حاضر بیشترین کاربرد را در بین روش های تعیین توالی دارد. اما روشهای تعیین توالی طولانی دیگری نیز وجود دارد که قابل توجه است ، مانند:
مزایا و معایب این فناوری ها را می توان در جدول زیر بررسی کرد:
دوره کارآموزی بیوانفورماتیک آزمایشگاه ژنیران