


خلاصهای از کم خونی آپلاستیک اکتسابی (Acquired Aplastic Anemia)
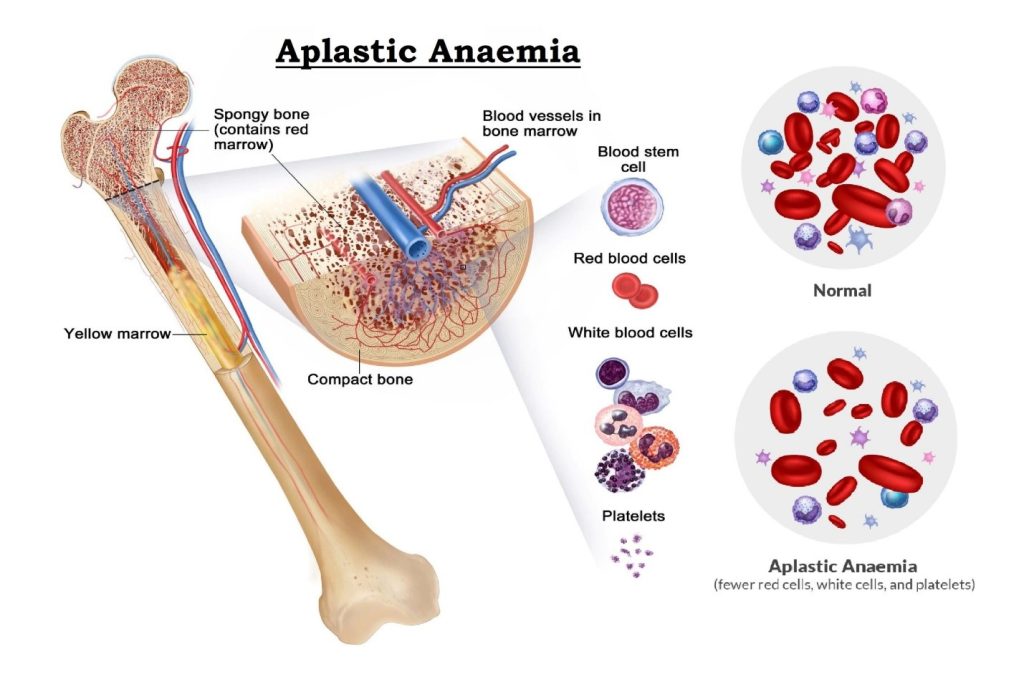
کم خونی آپلاستیک اکتسابی (Acquired Aplastic Anemia) یک اختلال خونی نادر و جدی است که به دلیل نارسایی مغز استخوان در تولید سلولهای خونی است. مغز استخوان (bone marrow) ماده اسفنجی است که در مرکز استخوانهای بدن، در بزرگسالان عمدتاً ستون فقرات، لگن و استخوانهای بزرگ پاها یافت میشود.
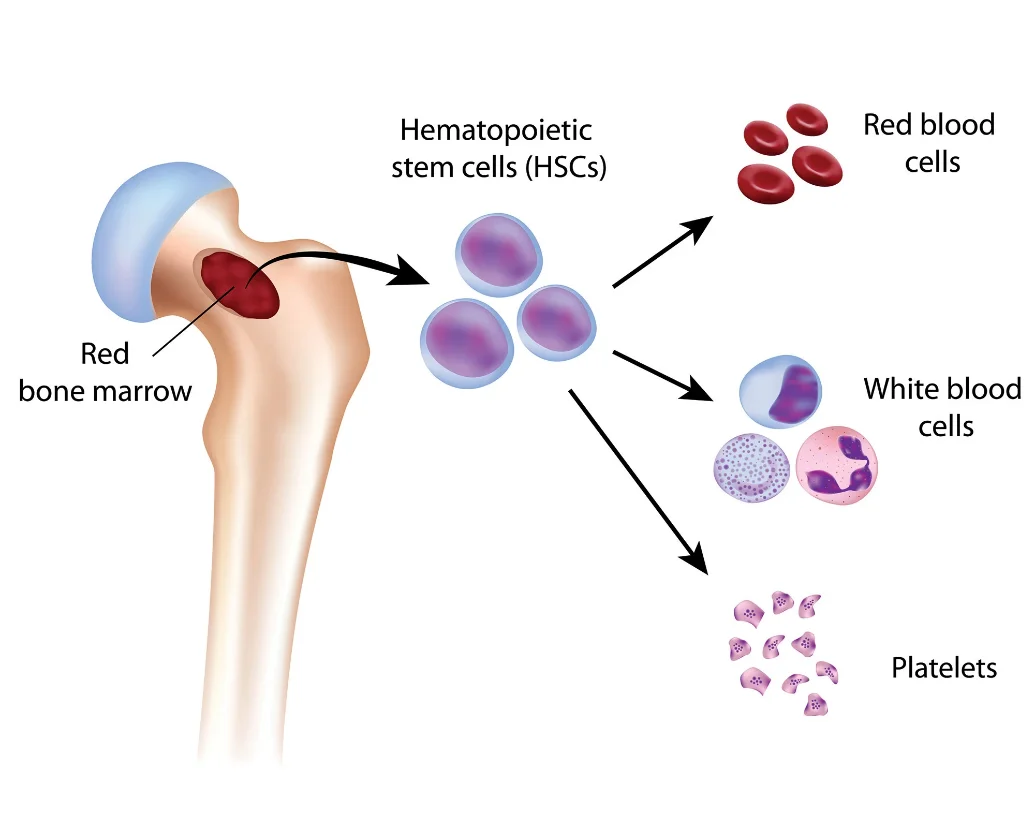
مغز استخوان حاوی سلولهای بنیادی خونساز است. سلولهای بنیادی میتوانند سلولهای بنیادی بیشتری تولید کنند (خود نوسازی یا self-renewal) و همچنین تمایز و تکثیر میشوند و گلبولهای قرمز (اریتروسیتها یا erythrocytes)، گلبولهای سفید (لکوسیتها یا leukocytes) و پلاکتها را تشکیل میدهند.
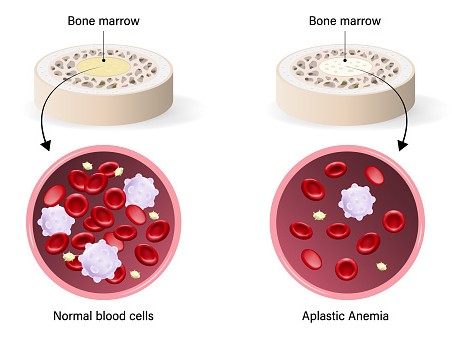
در کم خونی آپلاستیک اکتسابی، فقدان تقریباً کامل سلولهای بنیادی خونساز منجر به کاهش سطح گلبولهای قرمز و سفید و پلاکتها میشود (پانسیتوپنی یا pancytopenia). علائم کم خونی آپلاستیک علائم کم خونی، خونریزی و عفونت است.
اگرچه نارسایی مغز استخوان میتواند ثانویه به سایر اختلالات رخ دهد، اکثر کم خونی آپلاستیک اکتسابی به این دلیل است که سیستم ایمنی به اشتباه مغز استخوان را هدف قرار میدهد (بیماری خود ایمنی). در واقع، اکثر بیماران میتوانند به درمانی که سیستم ایمنی را سرکوب میکند، معمولاً ATG و سیکلوسپورین (cyclosporine)، پاسخ دهند.
معرفی کم خونی آپلاستیک اکتسابی
کم خونی آپلاستیک بر اساس شمارش خون به عنوان حالتی شدید طبقه بندی میشود. بیشتر بحثی که در ادامه میآید به کم خونی آپلاستیک شدید مربوط میشود. بیمارانی که شمارش خون آنها به طور متوسط کاهش یافته است. ممکن است نیاز به درمان نداشته باشد. علاوه بر این، برخی از کم خونی آپلاستیک که به صورت ژنتیکی به ارث میرسد، ممکن است ابتدا در بزرگسالی ظاهر شود، گاهی اوقات بدون سابقه خانوادگی بیماری خونی.
مترادفها
- کم خونی آپلاستیک ایدیوپاتیک (idiopathic aplastic anemia)
- کم خونی آپلاستیک ایمنی (immune aplastic anemia)
علائم و نشانههای کم خونی آپلاستیک اکتسابی
علائم کم خونی آپلاستیک اکتسابی در نتیجه ناتوانی مغز استخوان در تولید سلولهای خونی کافی رخ میدهد. علائم خاص در هر مورد متفاوت است. برخی از افراد ممکن است علائم خفیفی داشته باشند که برای چندین سال ثابت میماند. برخی دیگر ممکن است علائم جدی داشته باشند که میتواند منجر به عوارض تهدید کننده زندگی شود.
گلبولهای قرمز و سفید و پلاکتها در مغز استخوان تشکیل میشوند. سلولها در جریان خون آزاد میشوند تا در سراسر بدن حرکت کنند و وظایف خاص خود را انجام دهند. گلبولهای قرمز اکسیژن را به اندامهای بدن میرسانند، گلبولهای سفید خون به مبارزه با عفونتها کمک میکنند و پلاکتها برای توقف خونریزی، لختههایی تشکیل میدهند. سطح پایین گلبولهای قرمز در گردش خون، کم خونی (anemia) نامیده میشود. سطح پایین گلبولهای سفید خون به عنوان لکوپنی (leukopenia) شناخته میشود. سطح پایین پلاکتها به عنوان ترومبوسیتوپنی (thrombocytopenia) شناخته میشود.
افراد مبتلا به کم خونی ممکن است احساس خستگی، افزایش نیاز به خواب، ضعف، سبکی سر، سرگیجه، تحریک پذیری، سردرد، رنگ پوست رنگ پریده، مشکل در تنفس و علائم قلبی مانند درد قفسه سینه را تجربه کنند. خطر ابتلا به عفونتهای باکتریایی و قارچی در افراد مبتلا به لکوپنی افزایش مییابد. افراد مبتلا به ترومبوسیتوپنی به دنبال کمترین آسیب و خونریزی خود به خود از لثه و بینی بیشتر مستعد کبودی هستند. زنان ممکن است افزایش خون قاعدگی داشته باشند. علائم به شدت کم خونی، لکوپنی و ترومبوسیتوپنی بستگی دارد.
برخی از افراد مبتلا به کم خونی آپلاستیک اکتسابی نیز به طور همزمان دارای اختلال دیگری به نام هموگلوبینوری شبانه حمله ای (paroxysmal nocturnal hemoglobinuria یا PNH) هستند. آپلاستیک اکتسابی و PNH رابطه نزدیکی دارند که به طور کامل توسط محققان درک نشده است. اعتقاد بر این است که PNH در شرایط کم خونی آپلاستیک اکتسابی خود ایمنی و نارسایی مغز استخوان ایجاد میشود.
افراد مبتلا به کم خونی آپلاستیک اکتسابی نیز در معرض خطر تبدیل آن به اختلال مشابه دیگری به نام میلودیسپلازی (myelodysplasia) هستند. در تعداد کمی از موارد، کم خونی آپلاستیک اکتسابی ممکن است در نهایت به سرطان خون مبتلا شود. PNH ناشی از یک نقص ژنتیکی اکتسابی است که بر ژن PIGA که محدود به سلولهای بنیادی مغز است، تأثیر میگذارد. جهشهای ژن PIGA باعث میشود سلولهای خونی نسبت به افزایش تخریب توسط پروتئین مکمل (complement)، پروتئین ایمنی خون، حساس شوند.
در حدود نیمی از بیماران مبتلا به کم خونی آپلاستیک، شواهدی از PNH در هنگام مراجعه وجود دارد که توسط فلوسایتومتری (flow cytometry) تشخیص داده میشود. علاوه بر این، بیمارانی که به دنبال درمان سرکوب کننده سیستم ایمنی پاسخ میدهند ممکن است با PNH بهبود یابند. تعداد کمی از بیماران MDS با مغز استخوان هیپوپلاستیک یا کم سلولی وجود دارد، همان طور که در کم خونی آپلاستیک اکتسابی دیده میشود. این شرایط اغلب با یکدیگر اشتباه میشوند، بنابراین مشخص نیست که آیا یکی به دیگری تبدیل میشود یا خیر.
علل کم خونی آپلاستیک اکتسابی
اکثر موارد کم خونی آپلاستیک اکتسابی بدون هیچ علت قابل شناسایی یا به دلایل ناشناخته (ایدیوپاتیک یا idiopathic) رخ میدهند. محققان بر این باورند که بیشتر به این دلیل است که سیستم ایمنی به اشتباه مغز استخوان را هدف قرار میدهد (بیماری خود ایمنی). اختلالات خود ایمنی زمانی ایجاد میشوند که دفاع طبیعی بدن در برابر ارگانیسمهای “خارجی” یا مهاجم به دلایل ناشناخته شروع به حمله به بافتهای سالم میکند. آزمایشهایی برای تأیید این موضوع در هر مورد فردی به آسانی در دسترس نیست اما شواهد زیادی برای حمایت از این مکانیسم بیماریزا وجود دارد.
مغز استخوان حاوی سلولهای بنیادی خونساز (hematopoietic stem cells) است. این سلولهای بنیادی میتوانند تقسیم شوند، تمایز پیدا کنند و به گلبولهای قرمز یا سفید یا پلاکت تبدیل شوند.
در کمخونی آپلاستیک، یک رویداد تسریع کننده فرض میشود که باعث تخریب سلولهای بنیادی خونساز با واسطه ایمنی میشود. اعتقاد بر این است که برخی از سلولهای سیستم ایمنی (لنفوسیتهای T) ابتداییترین سلولهایی را که میتوانند به سلولهای خونی تبدیل شوند، یعنی سلولهای بنیادی خونساز، هدف قرار داده و از بین میبرند. افراد مبتلا به کم خونی آپلاستیک سلولهای بنیادی کافی برای تولید سلولهای خونی بالغ ندارند. به نظر میرسد که مغز استخوان با چربی جایگزین شده است. افراد مبتلا در نهایت دچار کمبود گلبولهای قرمز و سفید و پلاکتها میشوند (پانسیتوپنی).
در گذشته، کم خونی آپلاستیک اکتسابی با عوامل محیطی مختلف، به ویژه بنزن (benzene) مرتبط بوده است. بنزن به طور مستقیم برای سلولهای مغز استخوان مضر است. ارتباط نارسایی مغز استخوان با سایر مواد شیمیایی، مانند آفت کشها یا حشره کشها، کمتر ثابت شده است.
استفاده از برخی داروهای پزشکی نیز به ندرت با کم خونی آپلاستیک همراه است، مانند هپاتیت غیر ویروسی (nonviral hepatitis). هر دو ممکن است پاسخ سیستم ایمنی را تحریک کنند که به اشتباه سلولهای بنیادی خونساز را از بین میبرد. با این حال، اکثر موارد کم خونی آپلاستیک اکتسابی هیچ محرک محیطی قابل شناسایی ندارند.
جمعیتهای آسیب دیده
کم خونی آپلاستیک اکتسابی مردان و زنان را تقریباً به تعداد مساوی مبتلا میکند. در بیشتر موارد کودکان بزرگتر، نوجوانان یا بزرگسالان جوان را تحت تاثیر قرار میدهد. بروز کم خونی آپلاستیک در اروپا و اسرائیل دو مورد جدید در بین 1 میلیون نفر در سال است. میزان بروز در آسیا دو یا سه برابر بیشتر است. میزان بروز دقیق در ایالات متحده ناشناخته است، اگرچه برخی منابع میگویند که تقریباً 500 تا 1000 مورد جدید کم خونی آپلاستیک در سال تشخیص داده میشود.
اختلالات با علائم مشابه
علائم اختلالات زیر میتواند مشابه علائم کم خونی آپلاستیک اکتسابی باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد:
سندرمهای میلودیسپلاستیک یا Myelodysplastic syndromes (میلودیسپلازی، MDS) یک گروه نادر از اختلالات خونی هستند که در نتیجه رشد نامناسب سلولهای خونی در مغز استخوان رخ میدهند. سه نوع اصلی گلبولهای خون (یعنی گلبولهای قرمز، گلبولهای سفید و پلاکتها) تحت تأثیر قرار میگیرند. گلبولهای قرمز اکسیژن را به بدن میرسانند، گلبولهای سفید به مبارزه با عفونتها کمک میکنند و پلاکتها به لخته شدن کمک میکنند تا از دست دادن خون جلوگیری کنند. این سلولهای خونی که به طور نامناسب توسعه یافته اند به طور طبیعی رشد نمیکنند و وارد جریان خون میشوند.
در نتیجه، افراد مبتلا به MDS سطح سلولهای خونی به طور غیر طبیعی پایین (شمارش خون پایین یا low blood counts) دارند. گاهی اوقات تشخیص کم خونی آپلاستیک اکتسابی از MDS دشوار است. در MDS، بیماران اغلب به دلیل نارسایی مغز استخوان در تولید سلولهای خونی رنج میبرند و برخی از MDS میتوانند به لوسمی حاد تبدیل شوند. در MDS، اغلب ناهنجاریهای معمولی کروموزومهای سلولهای مغز و یا جهشهای مضر در ژنهای خاص موجود در ساقه خون ساز وجود دارد.
هموگلوبینوری پاروکسیسمال شبانه (Paroxysmal nocturnal hemoglobinuria یا PNH) یک اختلال نادر و اکتسابی سلولهای بنیادی است. یافته کلاسیک تخریب زودرس گلبولهای قرمز خون (همولیز یا hemolysis) است که منجر به دفعات مکرر هموگلوبین در ادرار (هموگلوبینوری یا hemoglobinuria) میشود. هموگلوبین رنگدانه قرمز و غنی از آهن خون است. افراد مبتلا به هموگلوبینوری ممکن است ادرار تیره رنگ یا خونی از خود نشان دهند.
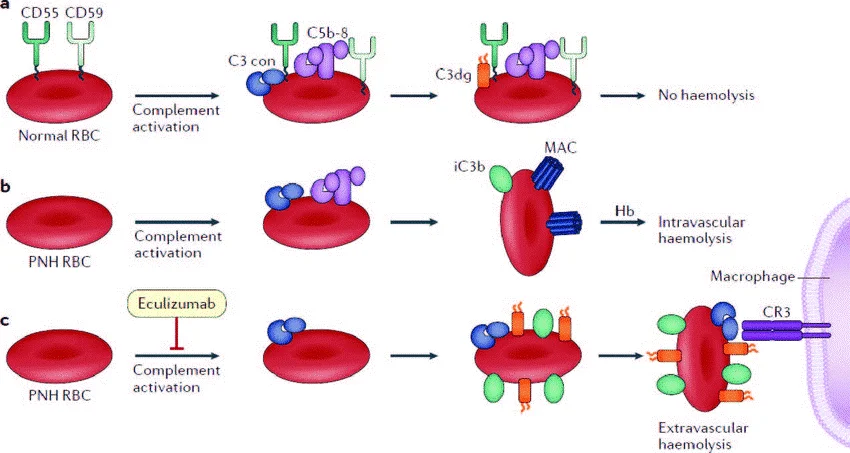
این یافته در صبح، پس از غلیظ شدن ادرار در طول شب هنگام خواب، برجستهتر است. علاوه بر همولیز، افراد مبتلا به PNH نیز مستعد ایجاد لختههای خونی مکرر و بالقوه تهدید کننده زندگی (ترومبوز یا thromboses) هستند. افراد مبتلا همچنین دارای درجاتی از اختلال عملکرد مغز استخوان هستند. اختلال شدید عملکرد مغز استخوان به طور بالقوه منجر به کاهش سطح گلبولهای قرمز و سفید خون و پلاکتها میشود (پانسیتوپنی). علائم خاص PNH بسیار متفاوت است و افراد مبتلا معمولاً همه علائم بالقوه مرتبط با این اختلال را نشان نمیدهند.
دو عامل برای ایجاد PNH ضروری است: یک جهش جسمی اکتسابی (که به کودکان منتقل نمیشود) ژن PIG-A که سلولهای بنیادی خونساز را تحت تأثیر قرار میدهد و سلولهای خونی “PNH” معیوب را ایجاد میکند و استعداد برای تکثیر و گسترش این سلولهای بنیادی معیوب PNH در شرایط نارسایی خود ایمنی مغز استخوان ایجاد میشود. محققان بر این باورند که سلولهای بنیادی معیوب PNH از حمله نادرست سیستم ایمنی جان سالم به در میبرند و تکثیر میشوند، در حالی که سلولهای بنیادی سالم از بین میروند و در نتیجه PNH ایجاد میشود.
کم خونی آپلاستیک همچنین ممکن است به عنوان بخشی از یک اختلال ارثی مانند کم خونی فانکونی (Fanconi anemia)، بیماریهای تلومر (telomere diseases)، سندرم شواچمن-دیاموند (Schwachman-Diamond syndrome)، سندرم آتاکسی-پانسیتوپنی (ataxia-pancytopenia syndrome) و غیره رخ دهد.
کم خونی فانکونی (Fanconi anemia) یک اختلال ژنتیکی نادر است که ممکن است در بدو تولد یا در دوران کودکی آشکار شود. در برخی موارد، کم خونی فانکونی ممکن است تا بزرگسالی تشخیص داده نشود. بسیاری از ژنهای مختلف بهعنوان جهش یافته در کم خونی فانکونی شناسایی شده اند و به طور کلی توانایی سلولها در ترمیم آسیب کروموزومی را دارند و مستعد آسیب به سلولهای بنیادی و در نهایت تبدیل لوسمیک (leukemic transformation) هستند.
این اختلال با کمبود تمام عناصر مغز استخوان از جمله گلبولهای قرمز، گلبولهای سفید و پلاکتها (پانسیتوپنی) مشخص میشود. کم خونی فانکونی همچنین ممکن است با ناهنجاریهای قلبی (cardiac)، کلیوی (renal) و یا اسکلتی و همچنین تغییر رنگهای لکه دار و قهوه ای (تغییرات رنگدانه) پوست همراه باشد.
چندین زیرگروه مختلف (گروههای مکمل) کم خونی فانکونی وجود دارد که تصور میشود هر کدام از آنها ناشی از تغییرات غیر طبیعی (جهش) در ژنهای مختلف است. به نظر میرسد هر زیرگروه علائم و یافتههای مشخصه یکسانی دارد (فنوتیپ). کم خونی فانکونی دارای توارث اتوزومال مغلوب است.
بیماریهای تلومر (telomere diseases) یا تلومروپاتیها (telomeropathies) نیز میتوانند منجر به کم خونی آپلاستیک شوند. در این شرایط ارثی، جهشهای ارثی در ژنهایی وجود دارد که انتهای کروموزومها را حفظ میکنند که تلومر (telomeres) نامیده میشود. انتهای کروموزوم به طور طبیعی با پیری سالم سلولها و ارگانیسمها فرسایش مییابد اما ساییدگی در بیماریهای تلومر تسریع میشود.
مانند کم خونی فانکونی، بیماران ممکن است تا بزرگسالی علائم بیماری را نشان ندهند. علاوه بر نارسایی مغز استخوان، تلوروپاتیها میتوانند به فیبروز ریوی و سیروز کبدی نیز منجر شوند. اعضای خانواده ممکن است دارای اندامهای مختلف و یا چندین اندام آسیب دیده باشند و تظاهرات آن میتواند از خفیف تا شدید متغیر باشد.
تشخیص کم خونی آپلاستیک اکتسابی
تشخیص کم خونی آپلاستیک اکتسابی ممکن است زمانی مشکوک باشد که یک فرد سالم دارای سطوح پایین هر سه نوع سلول خونی باشد (پانسیتوپنی).
تشخیص ممکن است با ارزیابی بالینی کامل، شرح حال دقیق بیمار و انواع آزمایشات تخصصی، از جمله بیوپسی مغز استخوان (bone marrow biopsy)، تایید شود. در طی این روش، نمونه کوچکی از بافت مغز استخوان معمولاً از لگن (pelvis) با جراحی برداشته میشود و زیر میکروسکوپ بررسی میشود. در کم خونی آپلاستیک اکتسابی این نمونه کاهش چشمگیر یا فقدان کامل سلولی را نشان میدهد. ممکن است برای رد سایر اختلالات مانند لوسمی و تعیین اینکه آیا علت ارثی یا ژنتیکی وجود دارد، آزمایشات اضافی لازم باشد.
درمان کم خونی آپلاستیک اکتسابی
روشهای درمانی
درمان کم خونی آپلاستیک اکتسابی بسته به سن فرد، سلامت عمومی و شدت کم خونی آپلاستیک متفاوت است. هدف درمان اصلاح نارسایی مغز استخوان و همچنین درمان علائم و نشانههای فوری بیمار است. دو شکل اصلی درمان اختصاصی پیوند مغز استخوان و درمانهای سرکوب کننده ایمنی هستند.
درمان اولیه کم خونی آپلاستیک اکتسابی ممکن است در جهت بهبود علائمی باشد که ممکن است در نتیجه شمارش خون پایین ایجاد شود. چنین درمانی شامل تزریق گلبول قرمز برای اصلاح کم خونی، تزریق پلاکت برای درمان یا جلوگیری از خونریزی جدی و آنتی بیوتیک برای درمان یا پیشگیری از عفونت است.
پیوند مغز استخوان (Bone marrow transplantation)، به ویژه پیوند آلوژنیک (allogeneic transplant)، درمان انتخابی در کودکان و بزرگسالان جوان تر است. با پیوند مغز استخوان آلوژنیک، سلولهای غیر طبیعی مغز استخوان فرد مبتلا با شیمی درمانی ریشه کن شده یا از بین میروند و با مغز سالم به دست آمده از اهدا کننده جایگزین میشوند.
مغز استخوان اهدا کننده با تزریق داخل وریدی سلولهای اهدا کننده به بدن بیمار پیوند میشود، جایی که به مغز استخوان بیمار میرود و در نهایت شروع به تولید سلولهای خونی جدید میکند. بهترین گزینه برای پیوند مغز استخوان یک دوقلو، خواهر یا برادر یا خویشاوند نزدیک یکسان است که بیشتر ساختار ژنتیکی بیمار را دارد. با این حال، در بسیاری از موارد، جستجو برای یک اهدا کننده غیر مرتبط و همسان ضروری است، یا اخیراً یکی از اعضای خانواده تا حدی همسان اهدا کننده است.
رد پیوند (Graft rejection) و بیماری پیوند در مقابل میزبان (graft-versus-host disease) از عوارض بالقوه هر روش پیوند، از جمله پیوند مغز استخوان است. عوارض بیماری پیوند در مقابل میزبان از پیوند مغز استخوان ممکن است از خفیف تا تهدید کننده زندگی متغیر باشد. ممکن است از داروها برای پیشگیری یا درمان رد پیوند یا بیماری پیوند در مقابل میزبان استفاده شود.
افرادی که کاندید پیوند مغز استخوان نیستند، چه به دلیل سن بالا و چه به دلیل نداشتن اهدا کننده مناسب، معمولا با درمان سرکوب کننده سیستم ایمنی (immunosuppressive treatment) درمان میشوند.
در این حالت از داروها برای سرکوب فعالیت سیستم ایمنی استفاده میشود. از آن جایی که تصور میشود بسیاری از موارد کم خونی آپلاستیک اکتسابی ناشی از حمله اشتباه سیستم ایمنی فرد به مغز استخوان است، سرکوب فعالیت سیستم ایمنی اغلب به مغز استخوان اجازه میدهد تا بهبود یابد و در نهایت شروع به تولید سلولهای خونی جدید کند. دو عامل سرکوب کننده سیستم ایمنی که به تنهایی یا به صورت ترکیبی تجویز میشوند، عبارتند از آنتی تیموسیت گلوبولین (ATG یا antithymocyte globulin) و سیکلوسپورین (cyclosporine).
ATG اسب در درمان کم خونی آپلاستیک موثرتر از ATG خرگوش است.
درمان سرکوبکننده سیستم ایمنی میتواند شمارش خون فرد مبتلا را برای دورههای طولانی به سطح نرمال یا نزدیک به نرمال بازگرداند. با این حال، بهبود ممکن است دائمی نباشد و در صورت بروز عود کم خونی آپلاستیک، دوره درمان باید تکرار شود. علاوه بر این، افرادی که با موفقیت به درمان سرکوب کننده سیستم ایمنی پاسخ میدهند، همچنان در معرض خطر ابتلا به PNH، میلودیسپلازی یا لوسمی هستند.
تقریباً یک سوم افراد تحت درمان با داروهای سرکوب کننده سیستم ایمنی به درمان پاسخ نمیدهند (کم خونی آپلاستیک مقاوم یا refractory aplastic anemia). در این موارد، درمان با پیوند سلولهای بنیادی خونساز ممکن است در نظر گرفته شود. سرکوب سیستم ایمنی میتواند در کم خونی آپلاستیک مقاوم و همچنین برای بیمارانی که عود کرده اند تکرار شود.
فاکتورهای رشد خونساز مانند اریتروپویتین (erythropoietin) و نوپوژن (neupogen) در کم خونی آپلاستیک موثر نیستند اما به طور شگفت انگیزی eltrombopag، محرک تولید پلاکت، در بهبود شمارش خون در کم خونی آپلاستیک مقاوم موثر بود.
در سال 2014، Promacta برای درمان بیماران مبتلا به کم خونی آپلاستیک شدید که پاسخ کافی به درمان سرکوب کننده سیستم ایمنی نداشته اند و کاندیدای پیوند سلولهای بنیادی خونساز نیستند، تایید شد. هنگامی که eltrombopag با سرکوب ایمنی استاندارد به عنوان درمان خط اول ترکیب شد، میزان پاسخ و نتیجه کامل بیشتر از سرکوب سیستم ایمنی به تنهایی بود.
مطالب مرتبط با کم خونی آپلاستیک اکتسابی:
مترجم: فاطمه فریادرس