


اطلاعات عمومی,ویکی ژن
لینوکس و R در آنالیز داده های NGS

بخش اول: مبانی لینوکس
لینوکس چیست؟
لینوکس یک خانواده از سیستمعاملهای متنباز و رایگان است که بر پایهی هستهی لینوکس (Linux Kernel) ساخته شدهاست. هستهی لینوکس، که از سال ۱۹۹۱ توسط لینوس توروالدز توسعه داده شده، قلب این سیستمعامل است و به دلیل انعطافپذیری و تطبیقپذیری بالای آن، در زمینههای مختلفی از سرورهای وب گرفته تا گوشیهای هوشمند و حتی ابررایانهها استفاده میشود. برای درک بهتر این موضوع، بهتر است بدانید ۹۰٪ زیرساختهای ابری و ۷۴٪ از گوشیهای هوشمند جهان از لینوکس بهره میبرند.

لینوکس بهصورت توزیعهای مختلف (Distributions) یا بهاختصار Distros عرضه میشود که هر کدام ویژگیها و کاربردهای خاص خود را دارند. از جمله توزیعهای معروف میتوان به اوبونتو (Ubuntu)، (Debian) و (CentOS) اشاره کرد. این توزیعها، نسخههای سفارشیشدهای از لینوکس هستند که با نرمافزارها و تنظیمات مختلف برای نیازهای گوناگون طراحی شدهاند.
برخلاف سیستمعاملهایی مثل ویندوز یا مکاواس که معمولاً رابط گرافیکی (GUI) را بهعنوان روش اصلی تعامل با کاربر ارائه میدهند، لینوکس به شدت به رابط خط فرمان (Command Line Interface) وابسته است. این ویژگی به کاربران امکان میدهد تا از طریق ترمینال یا شل (Shell) دستورات متنی را وارد کنند و مستقیماً با سیستمعامل ارتباط برقرار کنند. این قابلیت بهویژه برای تحلیل دادههای پیچیده و سنگین، مانند دادههای توالییابی نسل بعد (NGS)، بسیار ارزشمند است.
ساختار فایلسیستم لینوکس
ساختار فایلسیستم لینوکس: مفاهیم دایرکتوری، Root و Home
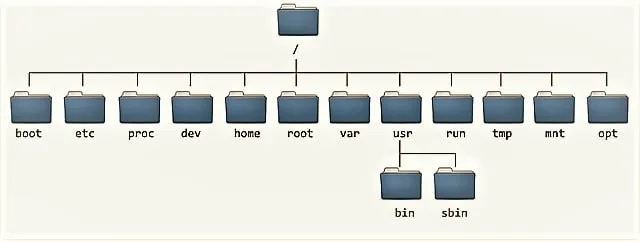
سیستمعامل لینوکس به دلیل ساختار منظم و سلسلهمراتبی فایلسیستم خود، محیطی قدرتمند برای مدیریت فایلها و اجرای برنامهها فراهم میکند. این ساختار ممکن است در ابتدا برای کاربران جدید پیچیده به نظر برسد، اما با شناخت مفاهیم اصلی آن، مانند دایرکتوریها، دایرکتوری ریشه (Root) و دایرکتوری خانگی (Home)، میتوان بهراحتی در آن حرکت کرد و از قابلیتهایش بهره برد. در این مقاله، نقش این مفاهیم را در تحلیل دادههای زیستی، بهویژه NGS، بررسی میکنیم.
- ساختار سلسلهمراتبی فایلسیستم لینوکس
فایلسیستم لینوکس مانند درختی سازماندهی شده که تمام فایلها و دایرکتوریها از یک نقطهی شروع به نام ریشه (Root) منشعب میشوند. این ساختار سلسلهمراتبی به کاربران اجازه میدهد فایلها و برنامهها را بهصورت منظم مدیریت کنند. دایرکتوریها (پوشهها) در لینوکس نقش کلیدی دارند و هر کدام برای هدف خاصی طراحی شدهاند. برای مثال:
- برخی دایرکتوریها مثل /bin برنامههای اجرایی ضروری را نگه میدارند.
- دایرکتوری /etc فایلهای پیکربندی سیستم را ذخیره میکند.
- دایرکتوری /home محل ذخیرهی فایلهای شخصی کاربران است.
در تحلیل NGS، این ساختار منظم به محققان کمک میکند تا دادههای بزرگ (مثل فایلهای FASTQ یا BAM) را در دایرکتوریهای مشخصی مثل /home یا /tmp سازماندهی کنند و ابزارهای بیوانفورماتیک را از دایرکتوریهایی مثل /bin یا /usr اجرا کنند.
- دایرکتوری ریشه (Root)
دایرکتوری ریشه با علامت / نشان داده میشود و نقطهی شروع تمام فایلها و دایرکتوریها در لینوکس است. این دایرکتوری مانند پایهی درخت فایلسیستم عمل میکند و شامل دایرکتوریهای مهمی است که برای عملکرد سیستم ضروریاند. ویژگیهای کلیدی دایرکتوری ریشه عبارتند از:
- محل دایرکتوریهای اصلی
دایرکتوریهایی مثل /bin (برای برنامههای اجرایی)، /etc (برای فایلهای پیکربندی)، /home (برای فایلهای کاربران) و /lib (برای کتابخانههای سیستمی) در زیرمجموعهی ریشه قرار دارند.
- فایلهای مخفی
برخی فایلها و دایرکتوریها در ریشه با نقطه (.) شروع میشوند (مثل .bashrc) و مخفی هستند. برای دیدن آنها، میتوانید از دستور ls -a استفاده کنید.
- اهمیت در مدیریت سیستم
برای مدیریت لینوکس، باید بدانید چه فایلهایی در ریشه قرار دارند و چگونه با دستوراتی مثل cd (تغییر دایرکتوری) و ls (نمایش محتویات) در آن حرکت کنید.
- کاربرد در NGS
در تحلیل NGS، دایرکتوری ریشه به شما کمک میکند تا ابزارهای سیستمی (مثل SAMtools در /bin) یا فایلهای پیکربندی (در /etc) را پیدا کنید. برای مثال، میتوانید تنظیمات یک ابزار بیوانفورماتیک را در /etc تغییر دهید تا عملکرد بهتری داشته باشد.
- دایرکتوری خانگی (Home)
دایرکتوری خانگی با علامت ~ نشان داده میشود و فضای شخصی هر کاربر در سیستم است. وقتی وارد لینوکس میشوید، بهطور خودکار به دایرکتوری خانگی خود هدایت میشوید. ویژگیهای کلیدی این دایرکتوری عبارتند از:
- فضای شخصی کاربر
هر کاربر دایرکتوری خانگی خودش را دارد (مثلاً /home/username) که میتواند در آن فایلها، اسکریپتها و دادههای شخصی را ذخیره کند.
- زیرمجموعههای پیشفرض
دایرکتوری خانگی معمولاً شامل زیرمجموعههایی مثل Documents، Downloads، Pictures و Videos است که برای سازماندهی فایلها طراحی شدهاند. مثلاً میتوانید دادههای خام NGS را در Documents ذخیره کنید.
- انعطافپذیری
کاربران میتوانند زیرمجموعههای دلخواه خود را بسازند. مثلاً میتوانید دایرکتوریای به نام ngs_data برای ذخیرهی فایلهای توالییابی ایجاد کنید.
- مدیریت آسان
با سازماندهی منطقی فایلها در دایرکتوری خانگی، پیدا کردن و استفاده از دادهها سریعتر میشود.
- کاربرد در NGS
دایرکتوری خانگی بهترین مکان برای ذخیرهی دادههای خام NGS، اسکریپتهای تحلیل (مثل اسکریپتهای Bash برای اجرای BWA) و نتایج پردازش است. مثلاً میتوانید فایلهای FASTQ را در /home/username/ngs_data ذخیره کنید و با ابزارهایی مثل FastQC در همان دایرکتوری تحلیل کنید.
اهمیت شناخت دایرکتوریها در تحلیل NGS
شناخت ساختار دایرکتوریهای لینوکس، بهویژه ریشه و خانگی، برای تحلیل NGS ضروری است، چون:
- مدیریت دادهها: دادههای NGS معمولاً حجم بالایی دارند و نیاز به سازماندهی دقیق در دایرکتوریهایی مثل /home یا /tmp دارند.
- اجرای ابزارها: بسیاری از ابزارهای بیوانفورماتیک (مثل GATK یا Bowtie2) در دایرکتوریهایی مثل /bin یا /usr نصب میشوند و باید بدانید کجا هستند.
- اتوماسیون: با استفاده از اسکریپتها در دایرکتوری خانگی، میتوانید فرآیندهای پیچیدهی NGS مثل همترازسازی یا فیلتر کردن دادهها را خودکار کنید.
- امنیت: تنظیم مجوزهای فایل (مثل استفاده از chmod در دایرکتوری خانگی) از دادههای حساس NGS محافظت میکند.
در پروژههای NGS، معمولاً دادههای خام، نتایج میانی و خروجیهای نهایی در دایرکتوری خانگی ذخیره میشوند، در حالی که ابزارهای سیستمی و پیکربندیها در ریشه یا زیرمجموعههای آن (مثل /bin یا /etc) قرار دارند. این تفکیک باعث میشود کار با دادهها و ابزارها منظمتر باشد.
یکی از ویژگیهای کلیدی لینوکس، ساختار فایلسیستم آن است که بر اساس استانداردFilesystem Hierarchy Standard (FHS) طراحی شده است. این استاندارد، دایرکتوریهای مختلف را با کاربردهای مشخص تعریف میکند:
- / (Root Directory)
نقطهی شروع فایل سیستم لینوکس است و تمام دایرکتوریها و فایلها زیر آن قرار دارند.
- /home/
دایرکتوری خانگی کاربران عادی را ذخیره میکند. برای مثال، اگر نام کاربری شما sammy باشد، دایرکتوری خانگی شما /home/sammy خواهد بود.
- /root/
دایرکتوری خانگی کاربر ریشه (Root User) است که دسترسی کامل به سیستم دارد.
- /etc/
فایلهای پیکربندی سیستم در این دایرکتوری ذخیره میشوند.
- /var/
دادههای متغیر، مانند لاگها و فایلهای موقت، در این دایرکتوری قرار دارند.
- /usr/
شامل نرمافزارها و فایلهای سیستمی است که کاربران میتوانند از آنها استفاده کنند.
این ساختار منظم به تحلیلگران NGS کمک میکند تا دادهها و ابزارهای خود را بهصورت سازمانیافته مدیریت کنند. برای مثال، میتوانید فایلهای دادهی NGS را در دایرکتوری خانگی خود ذخیره کنید و اسکریپتهای Bash را در یک دایرکتوری جداگانه نگه دارید.
دستورات پایه لینوکس برای شروع
برای شروع کار با لینوکس در تحلیل NGS، یادگیری چند دستور پایه ضروری است:
- pwd
نمایش مسیر دایرکتوری کنونی (مثال: /home/sammy).
- ls
نمایش لیست فایلها و دایرکتوریهای موجود در دایرکتوری کنونی.
- cd
تغییر دایرکتوری (مثال: cd /home/sammy/testdir1 برای رفتن به دایرکتوری testdir1).
- mkdir
ساخت دایرکتوری جدید (مثال: mkdir NGS_data برای ایجاد دایرکتوری به نام ngs_data).
- touch
ساخت فایل خالی (مثال: touch analysis.sh برای ایجاد یک فایل اسکریپت Bash).
- mv
جابهجایی یا تغییر نام فایل/دایرکتوری (مثال: mv file.txt newfile.txt).
- cp
کپی کردن فایل/دایرکتوری (مثال: cp data.fastq data_backup.fastq).
- rm
حذف فایل یا دایرکتوری (مثال: rm file.txt یا rm -r directory برای حذف دایرکتوری و محتویات آن).
- cat
نمایش محتوای یک فایل (مثال: cat file.txt).
- less
نمایش محتوای فایل بهصورت صفحهبهصفحه (مثال: less data.fastq).
- nano
ویرایشگر متنی ساده برای ویرایش فایلها (مثال: nano analysis.sh).
منبع
https://www.digitalocean.com/community/tutorials/an-introduction-to-linux-basics
چرا لینوکس برای تحلیل دادههای توالییابی نسل بعد (NGS) استفاده می گردد؟
تحلیل دادههای توالییابی نسل بعد (Next-Generation Sequencing یا NGS) یکی از پیچیدهترین و پرحجمترین فرآیندها در حوزهی بیوانفورماتیک است که نیازمند ابزارهای محاسباتی قدرتمند و انعطافپذیر است. لینوکس به دلیل ویژگیهای منحصربهفرد خود، بهعنوان سیستمعامل انتخابی برای بسیاری از زیست شناسان و تحلیلگران دادههای زیستی شناخته میشود. در زیر، دلایلی که لینوکس را برای تحلیل دادههای NGS مناسب میکنند آورده شده است:
- پشتیبانی از ابزارهای تخصصی بیوانفورماتیک: بسیاری از ابزارهای اصلی تحلیل دادههای زیستی، بهطور خاص برای محیط لینوکس طراحی و توسعه داده شدهاند. این ابزارها به دلیل بهینهسازی برای لینوکس، عملکرد بهتری در این سیستمعامل دارند. برای مثال، این ابزارها بهراحتی در ترمینال لینوکس اجرا میشوند و میتوانند فایلهای بزرگ دادههای NGS (مانند FASTQ یا BAM) را بهسرعت پردازش کنند.
- پشتیبانی از توسعه نرمافزار در زبانهای مختلف: لینوکس از زبانهای برنامهنویسی متعددی مانند Python، Perl، و R پشتیبانی میکند که در تحلیل دادههای زیستی بسیار پرکاربرد هستند. این انعطافپذیری به زیست شناسان اجازه میدهد تا اسکریپتها و ابزارهای سفارشی خود را برای تحلیل NGS بنویسند و اجرا کنند. برای مثال، میتوانید اسکریپتی در Bash یا Python بنویسید که فرآیند همترازسازی (Alignment) را خودکار کند.
- عدم نیاز به رابط گرافیکی (GUI): بسیاری از ابزارهای تحلیل NGS نیازی به رابط گرافیکی ندارند و از طریق خط فرمان اجرا میشوند. توسعهدهندگان این ابزارها معمولاً ترجیح میدهند نرمافزارهایی بسازند که بدون نیاز به محیط گرافیکی کار کنند، زیرا این کار سریعتر و سادهتر است. لینوکس با ارائهی محیط شل (Shell)، مانند Bash، این امکان را فراهم میکند تا کاربران دستورات را بهصورت متنی وارد کنند و فرآیندهای پیچیده را با چند خط کد مدیریت کنند.
- پشتیبانی از چندکاربره بودن و دسترسی از راه دور: لینوکس به چندین کاربر اجازه میدهد بهطور همزمان به سیستم وارد شوند و وظایف خود را انجام دهند. این ویژگی برای محیطهای تحقیقاتی، مانند سرورهای دانشگاهی یا ابررایانهها، بسیار مفید است. برای مثال، یک زیست شناس میتواند از طریق SSH به یک سرور لینوکسی متصل شود و تحلیلهای سنگین NGS را از راه دور انجام دهد، دقیقاً به همان صورتی که گویی پشت سیستم نشسته است. این قابلیت بهویژه در تیمهای تحقیقاتی که نیاز به همکاری دارند، بسیار ارزشمند است.
- قابلیت Multitasking: لینوکس بهخوبی از پس مدیریت چندین وظیفه بهطور همزمان برمیآید. در تحلیل NGS، ممکن است نیاز باشد چندین فرآیند مانند پیشپردازش دادهها، همترازسازی، و تجزیهوتحلیل بهصورت همزمان اجرا شوند. لینوکس با ابزارهایی مانند bg (برای اجرای وظایف در پسزمینه) و fg (برای آوردن وظایف به پیشزمینه) این امکان را فراهم میکند تا کاربران بهراحتی وظایف خود را مدیریت کنند. برای مثال، میتوانید یک اسکریپت تحلیل را در پسزمینه اجرا کنید و همزمان فایلهای دیگر را بررسی کنید.
- امنیت و کنترل دسترسی: دادههای NGS اغلب حساس هستند و ممکن است شامل اطلاعات ژنومی بیماران یا دادههای تحقیقاتی محرمانه باشند. لینوکس با ارائهی سیستم پیشرفتهی مدیریت مجوزها (Permissions) به کاربران اجازه میدهد تا دسترسی به فایلها و دایرکتوریها را دقیقاً کنترل کنند. بهعنوان مثال، با استفاده از دستور chmod، میتوانید مشخص کنید که فقط خودتان (کاربر) بتوانید فایلهای داده را بخوانید، ویرایش کنید یا اجرا کنید. این سطح از امنیت برای حفاظت از دادههای حساس NGS بسیار مهم است.
- مدیریت فایلهای بزرگ: دادههای NGS معمولاً حجم بسیار بالایی دارند (چند گیگابایت یا حتی ترابایت). لینوکس ابزارهای خط فرمان قدرتمندی مانند grep، awk، cut، و sort ارائه میدهد که برای پردازش و مدیریت این فایلها بهینه شدهاند. برای مثال، با دستور grep میتوانید بهسرعت توالیهای خاصی را در یک فایل FASTQ جستجو کنید، یا با دستور sort دادهها را مرتب کنید.
- انتقال فایلها با سهولت: لینوکس ابزارهایی مانند wget و scp را برای دانلود و انتقال فایلها فراهم میکند. برای مثال، میتوانید با دستور wget فایلهای دادهی NGS را از یک سرور آنلاین دانلود کنید یا با scp فایلها را بین کامپیوتر شخصی و سرور تحقیقاتی منتقل کنید. این ابزارها بهویژه برای محققینی که نیاز به انتقال دادههای بزرگ بین سیستمها دارند، بسیار کاربردی هستند.
- پشتیبانی از سرورهای وب و ابررایانهها: بسیاری از سرورهای وب و ابررایانههایی که برای تحلیل NGS استفاده میشوند، از لینوکس بهره میبرند. این سیستمعامل به دلیل پایداری و کارایی بالا، انتخاب اول برای این نوع محیطها است. همچنین، بسیاری از برنامههای تحت وب که برای تحلیل دادههای زیستی استفاده میشوند، روی سرورهای لینوکسی میزبانی میشوند.
- جامعهی بزرگ و منابع آموزشی: لینوکس از یک جامعهی فعال و گسترده از کاربران و توسعهدهندگان برخوردار است. اگر در حین تحلیل دادههای NGS به مشکلی برخورد کنید، منابع زیادی مانند مستندات آنلاین، انجمنهای پرسشوپاسخ، و صفحات راهنما (man pages) در دسترس هستند. برای مثال، با اجرای دستور man grep میتوانید اطلاعات کاملی دربارهی نحوهی استفاده از این ابزار دریافت کنید.
مقایسه لینوکس با سایر سیستمعاملها (ویندوز و مک) در تحلیل زیستی

تحلیل زیستی، بهویژه تحلیل دادههای NGS، نیازمند سیستمعاملهایی است که بتوانند حجم عظیمی از دادهها را بهطور کارآمد پردازش کنند، ابزارهای تخصصی بیوانفورماتیک را پشتیبانی کنند و محیطی امن و انعطافپذیر ارائه دهند. در این بخش، لینوکس را با ویندوز و مکاواس (macOS) از منظر کاربرد در تحلیل زیستی مقایسه میکنیم.
-
لینوکس در تحلیل زیستی
ویژگیهای کلیدی لینوکس که آن را برای تحلیل زیستی مناسب میکنند عبارتند از:
- متنباز بودن: لینوکس رایگان است و کد منبع آن در دسترس همه قرار دارد، که امکان بررسی و اصلاح آن را برای رفع نیازهای خاص فراهم میکند.
- امنیت بالا: به دلیل نظارت گسترده جامعهی جهانی توسعهدهندگان، لینوکس کمتر در معرض نقصهای امنیتی و بدافزارها قرار دارد.
- پشتیبانی از ابزارهای بیوانفورماتیک: بسیاری از ابزارهای تحلیل NGS مانند BLAST، SAMtools، BWA و GATK بهطور خاص برای لینوکس طراحی شدهاند.
- مدیریت منابع: لینوکس به دلیل عدم نیاز به رابط گرافیکی سنگین، منابع سیستمی را برای پردازش دادههای بزرگ آزاد میکند.
- انعطافپذیری در سرورها: لینوکس در سرورها و ابررایانهها (که ۱۰۰٪ از ۵۰۰ ابررایانهی برتر جهان را تشکیل میدهند) بسیار پایدار و قدرتمند است.
-
ویندوز در تحلیل زیستی
ویژگیها و چالشهای ویندوز در حوزهی تحلیل زیستی عبارتند از:
- مزایا:
- رابط کاربری ساده: ویندوز با منوی استارت و قابلیتهایی مثل Task View، برای کاربرانی که با خط فرمان آشنا نیستند، راحتتر است.
- پشتیبانی گسترده از نرمافزارها: ویندوز طیف وسیعی از برنامهها را پشتیبانی میکند، از جمله برخی ابزارهای تحلیل زیستی مانند Geneious یا CLC Genomics Workbench.
- سازگاری با سختافزارهای متنوع: ویندوز روی انواع دستگاهها از لپتاپهای ارزان تا سیستمهای قدرتمند قابل اجراست.
- معایب:
- امنیت پایینتر: به دلیل محبوبیت گسترده، ویندوز هدف اصلی بدافزارها، ویروسها و حملات ransomware است. این موضوع برای دادههای حساس NGS که نیاز به حفاظت دارند، یک چالش بزرگ است.
- عملکرد ضعیف با فایلهای بزرگ: ویندوز برای پردازش فایلهای بزرگ (مانند فایلهای FASTQ یا BAM) بهینه نیست و ابزارهای خط فرمان آن (مانند PowerShell) به اندازهی Bash لینوکس قدرتمند نیستند.
- ناسازگاری با ابزارهای بیوانفورماتیک: بسیاری از ابزارهای تخصصی NGS برای لینوکس طراحی شدهاند و ممکن است در ویندوز نیاز به تنظیمات اضافی یا شبیهسازهایی مثل WSL (Windows Subsystem for Linux) داشته باشند.
- هزینه: ویندوز رایگان نیست و نیاز به خرید لایسنس دارد، که میتواند برای تیمهای تحقیقاتی با بودجه محدود مشکلساز باشد.
-
macOS در تحلیل زیستی
مکاواس، سیستمعامل شرکت اپل، بر پایهی یونیکس ساخته شده و از سال ۱۹۹۷ (پس از خرید NeXT توسط اپل) بهعنوان یک سیستمعامل قدرتمند و امن شناخته میشود. macOS به دلیل رابط کاربری زیبا و سازگاری با سختافزارهای اپل، در میان متخصصان خلاق و برخی محققان زیستی محبوب است. ویژگیها و چالشهای macOS در تحلیل زیستی عبارتند از:
- مزایا:
- پایهی یونیکس: مکاواس به دلیل ریشههای یونیکسی، با بسیاری از ابزارهای خط فرمان لینوکس (مانند Bash) سازگار است و میتواند برخی ابزارهای NGS را بدون نیاز به تغییرات زیاد اجرا کند.
- امنیت بالا: macOS به دلیل تعداد کمتر کاربران در مقایسه با ویندوز، کمتر مورد هدف بدافزارها قرار میگیرد. ویژگیهایی مثل Screen Time و پارتیشنبندی جداگانه برای سیستم و دادههای کاربر، امنیت را افزایش میدهند.
- رابط کاربری بصری: macOS با برنامههایی مثل Preview، iMovie و GarageBand، تجربهی کاربری سادهای ارائه میدهد که برای محققانی که به رابط گرافیکی عادت دارند، جذاب است.
- پشتیبانی از توسعهدهندگان: macOS به دلیل محیط توسعهی Cocoa و پشتیبانی از AppleScript، برای برنامهنویسان زیستی که میخواهند ابزارهای سفارشی بسازند، مناسب است.
- معایب:
- هزینهی بالا: سختافزارهای اپل (مانند MacBook Air از ۹۹۹ دلار یا iMac از ۱۲۹۹ دلار) گرانقیمت هستند، که میتواند برای تیمهای تحقیقاتی با بودجه محدود چالشبرانگیز باشد.
- محدودیت سختافزاری: مکاواس فقط روی دستگاههای اپل اجرا میشود و امکان سفارشیسازی سختافزار (مانند افزودن کارتهای گرافیک قدرتمند) محدود است، که برای تحلیلهای سنگین NGS مشکلساز میشود.
- ناسازگاری با برخی ابزارها: اگرچه macOS یونیکسمحور است، برخی ابزارهای تخصصی NGS ممکن است به تنظیمات اضافی نیاز داشته باشند یا بهخوبی در لینوکس اجرا نشوند.
- وابستگی به اکوسیستم اپل: macOS در اکوسیستم بستهی اپل عمل میکند، که ممکن است برای کاربرانی که به انعطافپذیری لینوکس عادت دارند، محدودکننده باشد.
-
مقایسهی مستقیم در تحلیل زیستی
برای درک بهتر تفاوتهای این سه سیستمعامل در تحلیل زیستی، آنها را از چند منظر کلیدی مقایسه میکنیم:
پشتیبانی از ابزارهای بیوانفورماتیک:
- لینوکس: بهترین پشتیبانی را برای ابزارهای NGS مانند SAMtools، BWA، GATK و Bioconductor ارائه میدهد، زیرا اکثر این ابزارها برای لینوکس طراحی شدهاند. محیط خط فرمان Bash امکان خودکارسازی وظایف را فراهم میکند.
- ویندوز: پشتیبانی محدودی دارد و بسیاری از ابزارها نیاز به شبیهسازهایی مثل WSL یا Cygwin دارند. برخی نرمافزارهای تجاری مانند Geneious در ویندوز اجرا میشوند، اما برای تحلیلهای سنگین مناسب نیستند.
- macOS: به دلیل پایهی یونیکسی، برخی ابزارهای لینوکس را پشتیبانی میکند، اما ممکن است نیاز به تنظیمات اضافی داشته باشد. نرمافزارهایی مثل Geneious در مکاواس نیز در دسترس هستند.
امنیت دادههای حساس:
- لینوکس: به دلیل نظارت جامعهی متنباز، کمتر در معرض بدافزارها و نقصهای امنیتی است. سیستم مجوزهای پیشرفته (chmod، chown) امکان مدیریت دقیق دسترسی به دادههای NGS را فراهم میکند.
- ویندوز: به دلیل محبوبیت بالا، هدف اصلی بدافزارها و ransomware است. Windows Defender ابزارهای امنیتی خوبی ارائه میدهد، اما برای دادههای حساس کافی نیست.
- macOS: امنیت بالایی دارد و کمتر مورد هدف بدافزارها قرار میگیرد، اما همچنان در مقایسه با لینوکس آسیبپذیرتر است.
عملکرد در پردازش دادههای بزرگ:
- لینوکس: به دلیل عدم نیاز به رابط گرافیکی سنگین و ابزارهای خط فرمان قدرتمند، برای پردازش فایلهای بزرگ (مانند FASTQ یا BAM) بهینه است. همچنین در سرورها و ابررایانهها عملکرد عالی دارد.
- ویندوز: برای پردازش فایلهای بزرگ بهینه نیست و رابط گرافیکی آن منابع سیستمی را مصرف میکند.
- macOS: عملکرد خوبی دارد، اما به دلیل هزینهی بالای سختافزار و محدودیتهای سفارشیسازی، برای تحلیلهای سنگین مناسب نیست.
هزینه:
- لینوکس: کاملاً رایگان است و میتواند روی سختافزارهای ارزانقیمت (مانند Raspberry Pi با هزینهی ۸۰ دلار) اجرا شود.
- ویندوز: نیاز به خرید لایسنس دارد و هزینهی سختافزارهای سازگار متغیر است.
- macOS: نیاز به خرید سختافزارهای گرانقیمت اپل دارد، که هزینهی اولیه را بالا میبرد.
انعطافپذیری و سفارشیسازی:
- لینوکس: بالاترین سطح انعطافپذیری را ارائه میدهد. کاربران میتوانند توزیع، ابزارها و تنظیمات را به دلخواه خود سفارشی کنند.
- ویندوز: انعطافپذیری محدودی دارد و بیشتر برای کاربران عمومی طراحی شده است.
- macOS: در اکوسیستم بستهی اپل عمل میکند و سفارشیسازی محدود است.
جامعهی پشتیبانی و منابع آموزشی:
- لینوکس: جامعهی بزرگی از توسعهدهندگان و کاربران دارد که مستندات گسترده (مانند man pages) و انجمنهای آنلاین ارائه میدهند.
- ویندوز: پشتیبانی تجاری قوی از مایکروسافت و جامعهی کاربران عمومی دارد، اما منابع تخصصی برای تحلیل زیستی محدود است.
- macOS: پشتیبانی خوبی از اپل و جامعهی کاربران خلاق دارد، اما برای ابزارهای NGS به اندازهی لینوکس قوی نیست.
- منبع
https://ijaem.net/issue_dcp/A%20comparative%20study%20on%20Windows,%20macOS,%20and%20Linux.pdf
معرفی توزیعهای لینوکس مناسب برای NGS (Ubuntu، CentOS، Debian)
تحلیل دادههای NGS نیازمند محیطی پایدار، انعطافپذیر و بهینه برای پردازش حجم عظیمی از دادههای زیستی است. توزیعهای مختلف لینوکس (Linux Distributions) با بستهبندی هستهی لینوکس همراه با نرمافزارها و پیکربندیهای متنوع، گزینههای متعددی را برای محققان بیوانفورماتیک فراهم میکنند. انتخاب توزیع مناسب لینوکس میتواند تأثیر قابلتوجهی بر کارایی و بهرهوری تحقیقات NGS داشته باشد. در این بخش، دو توزیع محبوب لینوکس یعنی اوبونتو (Ubuntu) و (CentOS) را که برای تحلیل NGS مناسب هستند، معرفی و بررسی میکنیم.
-
اوبونتو (Ubuntu)
اوبونتو یکی از محبوبترین توزیعهای لینوکس است که به دلیل سادگی و راحتی استفاده، برای محققان تازهکار در حوزهی بیوانفورماتیک و تحلیل NGS بسیار مناسب است. این توزیع ویژگیهایی دارد که آن را به گزینهای ایدهآل تبدیل میکند:
- کاربری آسان: اوبونتو با رابط کاربری گرافیکی ساده (مثل GNOME) و فرآیند نصب راحت، برای کسانی که تجربهی کمی با لینوکس دارند، مناسب است. این ویژگی باعث میشود کاربران بهسرعت کار با سیستم را یاد بگیرند.
- مخزن نرمافزاری بزرگ: اوبونتو تعداد زیادی نرمافزار بیوانفورماتیک مثل SAMtools، BWA و GATK را در مخزن خود دارد. این ابزارها با دستورات سادهای مثل sudo apt install samtools بهراحتی نصب میشوند.
- نسخههای پایدار (LTS): نسخههای Long-Term Support (مثل Ubuntu 20.04 LTS) تا ۵ سال پشتیبانی میشوند و برای پروژههای تحقیقاتی که نیاز به نتایج ثابت و قابلاعتماد دارند، عالی هستند.
- پشتیبانی گسترده: اوبونتو جامعهی کاربری بزرگی دارد و منابع آموزشی زیادی مثل مستندات رسمی و انجمنهای آنلاین برای حل مشکلات در دسترس است.
- پشتیبانی از کانتینرها: اوبونتو از فناوریهای کانتینری مثل Docker و Singularity پشتیبانی میکند. این ابزارها به محققان کمک میکنند تا محیطهای یکسان و قابلتکرار برای تحلیل NGS بسازند.
چرا برای NGS مناسب است؟
اوبونتو به دلیل نصب آسان ابزارها و پایداری، برای پردازش فایلهای بزرگ NGS (مثل فایلهای FASTQ یا BAM) و اجرای خطوط لولهی بیوانفورماتیک بسیار مناسب است. برای مثال، میتوانید با نوشتن اسکریپتهای ساده در Bash، کارهایی مثل همترازسازی توالیها یا فیلتر کردن دادهها را خودکار کنید.
-
سنتاواس (CentOS)
سنتاواس به دلیل پایداری و طراحی برای محیطهای سازمانی، قبلاً یکی از گزینههای محبوب برای سرورها و تحلیلهای سنگین بود. از سال ۲۰۲۱، سنتاواس به CentOS Stream تغییر کرد که یک توزیع با بهروزرسانیهای مداوم است، اما Rocky Linux بهعنوان جایگزینی برای مدل سنتی سنتاواس معرفی شده است. ویژگیهای کلیدی سنتاواس برای تحلیل NGS عبارتند از:
- پایداری برای سرورها: سنتاواس بهگونهای طراحی شده که در محیطهای سروری و خوشههای محاسباتی (HPC) بسیار پایدار عمل کند. این برای تحلیلهای سنگین NGS که ساعتها یا روزها طول میکشند، بسیار مهم است.
- سازگاری با محیطهای ابری: بسیاری از سرورها و ابررایانههای مورد استفاده در تحلیل NGS از سنتاواس یا توزیعهای مشابه استفاده میکنند، که هماهنگی با این محیطها را آسان میکند.
- امنیت بالا: سنتاواس با بهروزرسانیهای امنیتی منظم، برای حفاظت از دادههای حساس NGS مناسب است.
- نصب ابزارهای NGS: اگرچه مخزن نرمافزاری سنتاواس به اندازهی اوبونتو گسترده نیست، ابزارهای NGS را میتوان با مدیر بستهی dnf یا بهصورت دستی (مثلاً از طریق کامپایل کد منبع) نصب کرد.
- جایگزین Rocky Linux: اگر به پایداری سنتی سنتاواس نیاز دارید، Rocky Linux گزینهای عالی است که همان مدل را دنبال میکند.
چرا برای NGS مناسب است؟
سنتاواس (یا Rocky Linux) برای تحلیلهای NGS در محیطهای سروری یا خوشههای محاسباتی که نیاز به پردازش موازی دادههای بزرگ دارند، مناسب است. برای مثال، میتوانید از آن برای اجرای ابزارهایی مثل Bowtie2 یا STAR روی خوشههای HPC استفاده کنید.
چرا این توزیعها برای NGS مناسباند؟
- اوبونتو: به دلیل سادگی، مخزن نرمافزاری بزرگ و نسخههای پایدار LTS، برای محققان تازهکار و پروژههایی که نیاز به راهاندازی سریع دارند، بهترین انتخاب است.
- سنتاواس (یا Rocky Linux): برای محیطهای سروری و خوشههای محاسباتی که تحلیلهای سنگین NGS انجام میدهند، مناسب است.
- نصب نرمافزار FastQC برای بررسی کیفیت دادههای NGS
FastQC ابزاری است که کیفیت دادههای توالییابی را بررسی میکند و گزارشهایی دربارهی مشکلات احتمالی (مثل آلودگی یا خوانشهای بیکیفیت) ارائه میدهد.
مراحل نصب:
به وبسایت FastQC (bioinformatics.babraham.ac.uk/projects/fastqc) بروید و فایل فشرده را دانلود کنید.
فایل را استخراج کنید، به دایرکتوری استخراجشده بروید و در ترمینال دستور زیر را اجرا کنید:
chmod 755 fastqc
این دستور فایل را قابلاجرا میکند.
یک Hard Link برای فایل fastqc در /usr/local/bin بسازید:
sudo ln /home/username/fastqc/fastqc /usr/local/bin
با دستور fastqc بررسی کنید که برنامه درست کار میکند.
کاربرد در NGS: FastQC به شما کمک میکند تا کیفیت دادههای خام (مثل فایلهای FASTQ) را قبل از پردازش بررسی کنید. وارد سرفصل کنترل کیفی شود
آشنایی با ترمینال لینوکس و محیط Shell
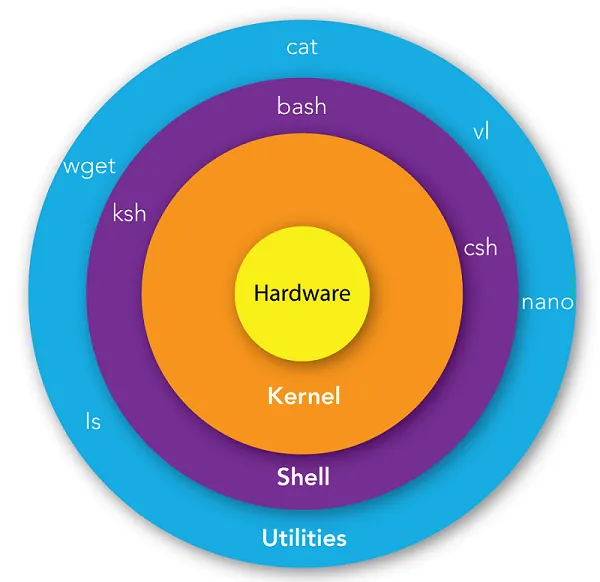
یکی از مهمترین ابزارهای لینوکس، ترمینال و محیط شل (Shell) است که به کاربران اجازه میدهد با سیستمعامل ارتباط برقرار کنند. در اینجا، مفاهیم پایهی ترمینال و شل را با زبانی ساده و رسمی توضیح میدهیم و کاربرد آنها در تحلیل NGS را بررسی میکنیم.
۱. کرنل چیست؟
کرنل هستهی اصلی سیستمعامل لینوکس است که مانند مغز سیستم عمل میکند و تمام منابع را مدیریت میکند، از جمله:
- مدیریت فایلها
- اجرای برنامهها
- مدیریت حافظه
- ارتباط با سختافزار
لینوس توروالدز، خالق لینوکس، فقط کرنل را توسعه داده و بقیهی سیستم لینوکس از ابزارها و کتابخانههای GNU و اسکریپتهای مدیریتی تشکیل شده است.
کاربرد در NGS: کرنل تضمین میکند که ابزارهای سنگین بیوانفورماتیک (مثل SAMtools یا Bowtie2) بهدرستی اجرا شوند و منابع سیستم بهینه استفاده شوند.
۲. شل چیست؟
شل برنامهای است که به کاربران اجازه میدهد با سیستمعامل ارتباط برقرار کنند. شل دستورات قابلفهم برای انسان (مثل ls یا cd) را دریافت و به زبانی تبدیل میکند که کرنل بفهمد. وقتی وارد ترمینال میشوید، شل بهطور خودکار فعال میشود.
شلها به دو نوع تقسیم میشوند:
- شل خط فرمان (Command Line Shell): کاربران دستورات را در ترمینال تایپ میکنند و نتیجه را بهصورت متنی میبینند.
- شل گرافیکی (Graphical Shell): رابط کاربری گرافیکی (GUI) مثل دسکتاپ اوبونتو که امکان باز کردن پنجرهها و کلیک روی آیکونها را فراهم میکند.
انواع شلهای خط فرمان:
- BASH (Bourne Again Shell)
رایجترین شل در لینوکس، با دستورات ساده و قدرتمند.
- CSH (C Shell)
شبیه زبان برنامهنویسی C.
- KSH (Korn Shell)
ترکیبی از ویژگیهای BASH و CSH
کاربرد در NGS: شل خط فرمان (مثل BASH) برای اجرای ابزارهای بیوانفورماتیک و خودکارسازی فرآیندهای تحلیل دادهها (مثل همترازسازی یا فیلتر کردن) ضروری است.
- ترمینال چیست؟
ترمینال برنامهای است که رابط متنی برای دسترسی به شل فراهم میکند. کاربران در ترمینال دستورات را تایپ میکنند و خروجیها را میبینند. برای باز کردن ترمینال در اوبونتو، کافی است در نوار جستجو کلمهی «Terminal» را تایپ کنید و آن را باز کنید.
ترمینال برای اجرای اسکریپتهای پیچیده یا دستورات ساده (مثل ls -l برای نمایش فایلها) استفاده میشود. کار با ترمینال برای مبتدیان ممکن است سخت باشد، چون نیاز به حفظ کردن دستورات دارد، اما با تمرین، بسیار قدرتمند و سریع میشود.
کاربرد در NGS: در ترمینال میتوانید ابزارهایی مثل FastQC را اجرا کنید تا کیفیت دادههای NGS را بررسی کنید یا اسکریپتهایی برای پردازش دادهها بنویسید.
مدیریت کاربران، مجوزها و امنیت دادههای حساس در لینوکس
مدیریت کاربران و مجوزها در سیستمعامل لینوکس یکی از مهمترین جنبههای تأمین امنیت سیستم و حفاظت از دادههای حساس، بهویژه در تحلیل دادههای NGS، محسوب میشود. لینوکس بهعنوان یک سیستمعامل چندکاربره، با استفاده از شناسه کاربری منحصربهفرد (UID) و شناسه گروه (GID)، دسترسی به فایلها، دایرکتوریها و منابع سیستمی را مدیریت میکند.
ابزارهایی مانند useradd برای ایجاد حساب کاربری جدید، usermod برای اصلاح ویژگیهای کاربر، userdel برای حذف کاربر، و groupadd برای ایجاد گروههای جدید به کار میروند. دستور passwd امکان تنظیم یا تغییر رمزعبور کاربران را فراهم میکند، در حالی که chmod برای تنظیم مجوزهای خواندن (r)، نوشتن (w) و اجرا (x) و chown برای تغییر مالکیت فایلها و دایرکتوریها استفاده میشود.
برای مثال، دستور chmod 640 file.txt به مالک فایل اجازه خواندن و نوشتن، به گروه اجازه خواندن، و به سایرین هیچ دسترسی نمیدهد. ابزار sudo به کاربران مجاز امکان اجرای دستورات با امتیازات بالاتر را میدهد، که با ویرایش فایل /etc/sudoers (با استفاده از visudo) میتوان دسترسیها را دقیقاً تنظیم کرد.
رعایت اصل حداقل دسترسی (Principle of Least Privilege)، که در آن کاربران تنها به منابع ضروری دسترسی دارند، خطر دسترسی غیرمجاز به دادههای حساس مانند فایلهای FASTQ یا BAM را کاهش میدهد. نظارت بر فعالیت کاربران با دستوراتی مانند last (برای بررسی تاریخچه ورودها)، who (برای مشاهده کاربران فعال)، و history (برای بازبینی دستورات اجرا شده) و استفاده از سیستم ممیزی auditd برای ثبت رویدادهای سیستمی، به شناسایی فعالیتهای مشکوک کمک میکند.
در تحلیل NGS، این ابزارها امکان حفاظت از دادههای ژنومی حساس و جلوگیری از نشت اطلاعات را فراهم میسازند.
بخش دوم
معرفی زبان R و نقش آن در تحلیل دادههای زیستی
زبان R یک محیط برنامهنویسی رایگان و متنباز است که برای انجام تحلیلهای آماری و ایجاد تصاویر گرافیکی توسعه یافته و به دلیل تواناییهایش در پردازش دادههای پیچیده، به ابزاری کلیدی در تحلیل دادههای زیستی، بهویژه دادههای ژنومی مانند NGS، تبدیل شده است. R به پژوهشگران امکان میدهد تا با استفاده از کدهای ساده، تحلیلهای عمیقی روی دادههای زیستی مانند توالیهای DNA، RNA یا دادههای پروتئینی انجام دهند.
این زبان بهگونهای طراحی شده که کاربران، حتی با دانش برنامهنویسی محدود، بتوانند دادههای خام را به اطلاعات علمی قابلفهم تبدیل کنند.
برای مثال، R میتواند به زیستشناسان کمک کند تا الگوهای بیان ژن در نمونههای بیمار و سالم را مقایسه کرده یا تغییرات ژنتیکی مرتبط با بیماریها را شناسایی کنند. RStudio، یک نرمافزار همراه برای R، محیطی کاربرپسند با ظاهری مرتب ارائه میدهد که کار با R را آسانتر میکند.
این محیط مثل یک دفتر کار دیجیتال است که ابزارهای نوشتن کد، دیدن نتایج، و مدیریت پروژهها را در یک جا جمع کرده و به پژوهشگران کمک میکند تا بدون سردرگمی، تحلیلهای خود را انجام دهند. RStudio با امکاناتی مثل نمایش همزمان کد و نتایج، نصب راحت این بستهها، و قابلیت همکاری تیمی از طریق اتصال به Git (یک سیستم برای بهاشتراکگذاری کدها بین محققان)، به کاربران اجازه میدهد بدون نیاز به دانش برنامهنویسی پیشرفته، روی تحلیل دادهها تمرکز کنند. این ابزارها، تحلیل دادههای زیستی را مثل مرتب کردن یک پازل پیچیده، ساده و قابلفهم میکنند.
یکی از مهمترین ویژگیهای R، پشتیبانی از بستههای تخصصی مانند Bioconductor است که مثل جعبهابزارهای اضافی برای R هستند و در بیوانفورماتیک بسیار محبوب است. این بستهها ابزارهایی برای بررسی دادههای ژنومی (مانند توالیهای DNA) یا دادههای پروتئینی ارائه میدهند.
این بستهها به کاربران اجازه میدهند تا وظایف پیچیدهای مانند تحلیل کیفیت توالیهای خواندهشده (Reads) در دادههای NGS، همترازسازی توالیها با ژنوم مرجع، یا شناسایی مسیرهای زیستی (Pathways) مرتبط با ژنهای خاص را انجام دهند. علاوه بر این، R از مخازن دیگری مانند CRAN نیز پشتیبانی میکند که شامل هزاران بسته برای تحلیلهای عمومی و زیستی است.
این تنوع در ابزارها باعث میشود R بتواند نیازهای مختلف پژوهشگران، از تحلیلهای آماری ساده تا مدلسازیهای پیشرفته، را برآورده کند.
R همچنین به دلیل قابلیتهای قوی در بصریسازی دادهها، نقش مهمی در ارائه نتایج علمی ایفا میکند. پژوهشگران میتوانند با استفاده از توابع پایه R یا بستههای پیشرفتهتر، نمودارهایی مانند هیستوگرام، نمودارهای پراکندگی یا نقشههای حرارتی (Heatmaps) تولید کنند که برای تفسیر دادههای ژنومی بسیار مفیدند.
برای مثال، یک نقشه حرارتی میتواند نشان دهد کدام ژنها در شرایط مختلف (مانند قبل و بعد از درمان) بیان متفاوتی دارند. این قابلیت بصریسازی، همراه با امکان اتصال R به پایگاههای داده مانند MySQL یا MongoDB، به پژوهشگران اجازه میدهد تا دادهها را از منابع متنوع جمعآوری کرده و بهصورت یکپارچه تحلیل کنند.
به این ترتیب، R نهتنها ابزاری برای تحلیل است، بلکه پلتفرمی برای مدیریت کل فرآیند تحقیق زیستی، از جمعآوری داده تا ارائه نتایج، به شمار میرود.
نقش R در بیوانفورماتیک فراتر از تحلیل دادههای خام است؛ این زبان به پژوهشگران کمک میکند تا پرسشهای زیستی پیچیده را با روشهای آماری و محاسباتی پاسخ دهند. برای نمونه، در تحلیل دادههای RNA-Seq، زبان R میتواند تفاوتهای آماری در بیان ژنها بین دو گروه (مانند بیماران سرطانی و افراد سالم) را با استفاده از روشهای آماری مانند آزمونهای فرضیه یا مدلسازی رگرسیون شناسایی کند. این تواناییها، همراه با انعطافپذیری R در کار با دادههای بزرگ و پیچیده، آن را به یکی از محبوبترین ابزارها در میان زیستفناوران تبدیل کرده است.
چرا R برای تحلیل دادههای NGS مناسب است؟ از دید RNA seq از این دید بازنویسی شود
یکی از مهمترین دلایل مناسب بودن R برای تحلیل دادههای NGS، پشتیبانی گسترده آن از بستههای تخصصی مانند Bioconductor است. این مخزن، که بهطور خاص برای بیوانفورماتیک طراحی شده، شامل ابزارهایی برای انجام وظایفی مانند ارزیابی کیفیت توالیها، نرمالسازی دادهها (مانند محاسبه RPKM برای RNA-Seq)، و تحلیل بیان تفریقی ژنها (Differential Gene Expression) است.
برای مثال، بسته edgeR در Bioconductor، که بر اساس توزیع منفی دوجملهای کار میکند، به کاربران اجازه میدهد تا ژنهایی که در شرایط مختلف (مانند بیماران سرطانی در مقایسه با افراد سالم) بیان متفاوتی دارند را شناسایی کنند. این ابزارها، که بهصورت رایگان در دسترس هستند، تحلیلهای پیچیده NGS را برای پژوهشگران با هر سطحی از مهارت برنامهنویسی ممکن میسازند.
R همچنین قابلیت انجام تحلیلهای آماری پیشرفته، مانند آزمونهای فرضیه (مانند آزمون خیدو یا t-test)، مدلسازی پیشبینیکننده (Predictive Modeling)، و machine learning را دارد که برای دادههای NGS بسیار کاربردیاند. برای نمونه، در تحلیل دادههای RNA-Seq، زبان R میتواند با استفاده از بستههایی مانند limma یا edgeR، ژنهای کلیدی مرتبط با بیماریها را شناسایی کرده و مسیرهای زیستی (Pathways) آنها را بررسی کند. این فرآیند به پژوهشگران کمک میکند تا درک بهتری از مکانیسمهای زیستی، مانند نقش ژنها در سرطان پروستات، بهدست آورند.
علاوه بر این، R امکان اتصال به پایگاههای داده عمومی مانند NCBI-GEO یا ArrayExpress را فراهم میکند، که به پژوهشگران اجازه میدهد دادههای خام NGS را مستقیماً وارد کرده و تحلیل کنند. این ویژگی، بهویژه برای آزمایشهای گرانقیمت NGS که تولید دادههای جدید ممکن است هزینهبر باشد، بسیار ارزشمند است.
همچنین، R با بستههایی مانند WGCNA (برای تحلیل شبکههای همبستگی) یا ابزارهای خوشهبندی، امکان بررسی شباهتها و تفاوتهای نمونهها را فراهم میکند، مثلاً برای شناسایی نمونههای غیرعادی (Outliers) در دادههای توالییابی.
این انعطافپذیری و دسترسی به ابزارهای متنوع، R را به گزینهای ایدهآل برای تحلیل دادههای NGS تبدیل کرده است، بهطوری که پژوهشگران میتوانند کل فرآیند تحلیل، از پیشپردازش تا تفسیر نهایی، را در یک محیط واحد انجام دهند.
نصب و راهاندازی R و محیط RStudio
R از طریق شبکه جامع آرشیو R (CRAN) توزیع میشود، و RStudio یک محیط توسعه یکپارچه (IDE) است که با ارائه رابط کاربری گرافیکی، کدنویسی و مدیریت پروژهها را آسانتر میکند. در این بخش راهنمای نصب این ابزارها روی سیستمعاملهای ویندوز، مک، و لینوکس را مشاهده کنید:
- نصب برای ویندوز
- دانلود و نصب R:
به وبسایت CRAN (https://cran.r-project.org) بروید و لینک «Download R for Windows» را انتخاب کنید. در صفحه بعدی، روی لینک «base» کلیک کنید، سپس لینک دانلود نسخه جدید R (مانند «Download R X.X.X for Windows») را انتخاب کنید.
فایل نصبی (.exe) را اجرا کنید. در طی مراحل نصب، گزینههای پیشفرض را تغییر ندهید. پس از نصب، میانبر R در منوی استارت ظاهر میشود.
- دانلود و نصب RStudio:
به وبسایت RStudio (https://posit.co/download/rstudio-desktop/) بروید و گزینه دانلود RStudio Desktop برای ویندوز را انتخاب کنید.
فایل نصبی را اجرا کنید و با تنظیمات پیشفرض ادامه دهید. پس از نصب، RStudio را از طریق آیکون دسکتاپ یا منوی استارت باز کنید.
- تأیید نصب:
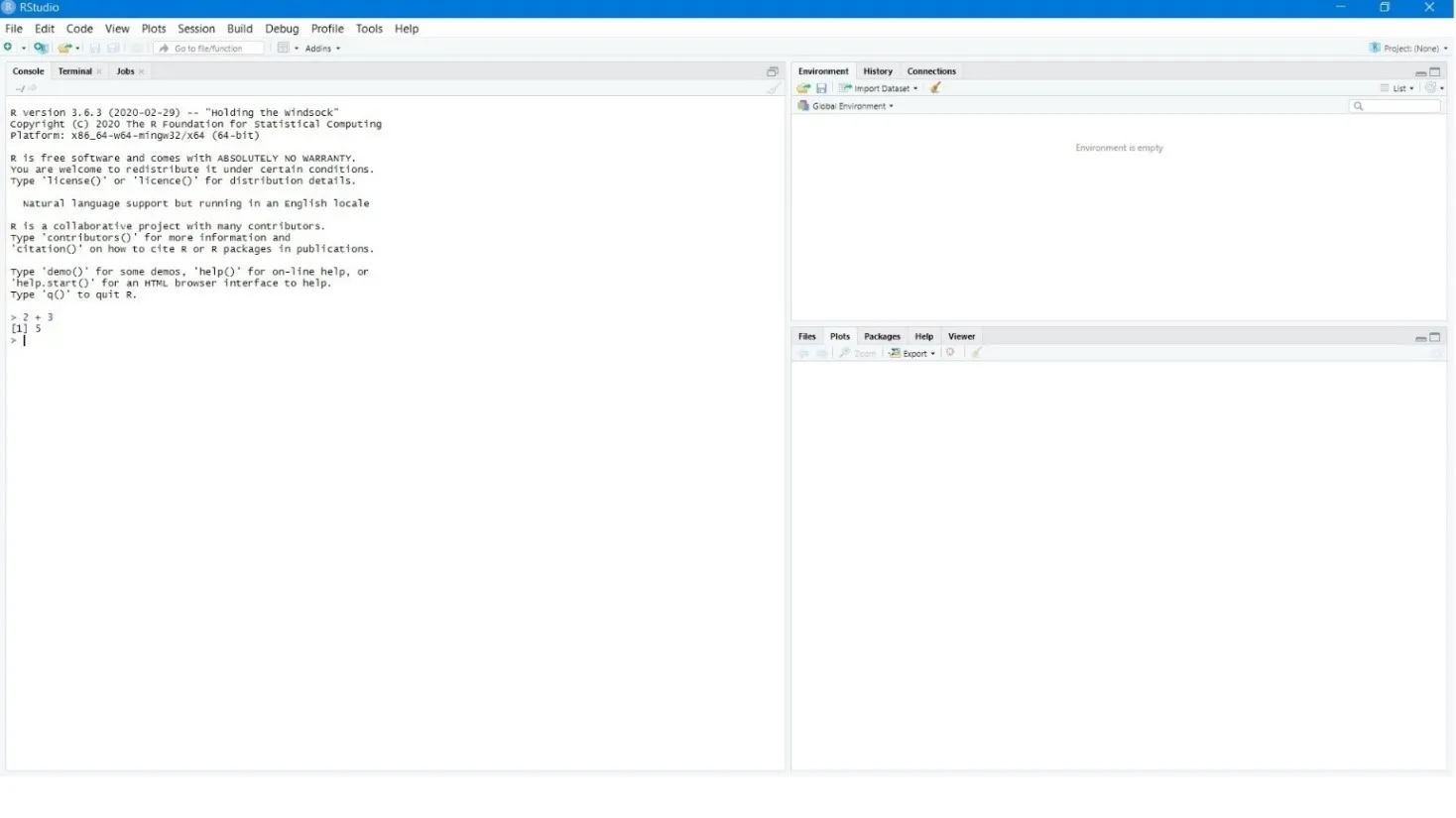
RStudio را اجرا کنید. پنجرهای با سه پنل ظاهر میشود: کنسول (برای اجرای کدها)، ویرایشگر متن، و پنلهای ابزار. در کنسول، دستور زیر را وارد کنید:
print(‘Hello world!’)
اگر خروجی “Hello world!” را ببینید، نصب موفقیتآمیز است.
- نصب برای مک
- دانلود و نصب R:
به وبسایت CRAN (https://cran.r-project.org) بروید و لینک «Download R for Mac» را انتخاب کنید. روی لینک بسته نصبی نسخه جدید R (مانند R-X.X.X.pkg) کلیک کنید.
فایل نصبی (.pkg) را اجرا کنید. تنظیمات پیشفرض برای اکثر کاربران مناسب است. اگر سیستم شما برای نصب نرمافزار جدید رمز عبور بخواهد، آن را وارد کنید.
- دانلود و نصب RStudio:
به وبسایت RStudio بروید و نسخه RStudio Desktop برای مک را دانلود کنید.
فایل نصبی را اجرا کنید و نصب را با تنظیمات پیشفرض تکمیل کنید.
- تأیید نصب:
RStudio را باز کنید و همان دستور print(‘Hello world!’) را در کنسول تست کنید. خروجی موفق نشاندهنده نصب صحیح است.
- نصب برای لینوکس
- نصب R:
بسیاری از توزیعهای لینوکس (مانند اوبونتو)، R را بهصورت پیشفرض دارند، اما برای اطمینان از نسخه جدید، به وبسایت CRAN و لینک «Download R for Linux» بروید. سپس، توزیع خود (مانند اوبونتو، دبیان، یا SUSE) را انتخاب کنید.
دستورالعملهای خاص هر توزیع در فایلهای README ارائه شده است. برای اوبونتو، معمولاً از دستورات زیر استفاده میشود:
sudo apt-get update
sudo apt-get install r-base
برخلاف ویندوز و مک که از فایلهای باینری (نصبی آماده) استفاده میکنند، در لینوکس معمولاً R از کد منبع ساخته میشود، که نیاز به دانش فنی بیشتری دارد.
نصب RStudio:
فایل نصبی RStudio برای لینوکس (مانند .deb برای اوبونتو) را از وبسایت RStudio دانلود کنید. برای اوبونتو، میتوانید از دستور زیر استفاده کنید:
sudo apt-get install gdebi-core
sudo gdebi rstudio-X.X.XXXX-amd64.deb
تأیید نصب:
RStudio را اجرا کنید (با تایپ rstudio در ترمینال یا از منوی برنامهها) و دستور print(‘Hello world!’) را تست کنید.
مفاهیم پایه در R: متغیرها، انواع داده و ساختارهای دادهای
برای بهرهبرداری مؤثر از زبان R، تسلط بر مفاهیم پایه شامل متغیرها، انواع داده، و ساختارهای دادهای ضروری است. این مفاهیم به پژوهشگران امکان میدهند دادهها را بهصورت منظم سازماندهی کرده و تحلیلهای پیشرفته انجام دهند. در این بخش به معرفی دقیق این مفاهیم میپردازیم.
-
متغیرها در R
متغیرها در R اشیایی هستند که برای ذخیره دادهها استفاده میشوند و بهعنوان ظرفهایی برای نگهداری اطلاعات عمل میکنند. برخلاف زبانهای برنامهنویسی مانند C که نیاز به تعریف صریح نوع متغیر دارند، در R میتوانید بهسادگی با استفاده از عملگر تخصیص <- مقداری را به یک نام اختصاص دهید. برای مثال:
name <- c(“Adam”, “Ben”, “Cecily”)
age <- c(21, 19, 20)
در اینجا، name و age متغیرهایی هستند که بهترتیب دادههای متنی (نام افراد) و عددی (سن) را ذخیره میکنند. این انعطافپذیری، بهویژه در تحلیل دادههای زیستی که ممکن است شامل انواع مختلفی از دادهها (مانند شناسههای ژن، مقادیر بیان، یا ویژگیهای بالینی) باشد، بسیار ارزشمند است. متغیرها میتوانند در هر زمان با تخصیص مقدار جدید تغییر کنند، و نوع آنها بهطور خودکار بر اساس محتوای جدید تنظیم میشود، که این ویژگی کار با دادههای پویا را سادهتر میکند.
-
انواع داده (Data Types)
هر متغیر در R دارای یک نوع داده یا mode است که ماهیت محتوای ذخیرهشده را تعیین میکند. شناخت انواع داده برای انتخاب ساختار مناسب و جلوگیری از خطاهای رایج در تحلیلهای آماری حیاتی است. انواع داده اصلی در R عبارتاند از:
- عددی (Numeric): شامل اعداد صحیح (Integer) یا اعشاری (Double)، مانند age <- c(21, 19, 20) یا مقادیر بیان ژن در دادههای RNA-Seq. این نوع برای محاسبات عددی، مانند میانگین یا انحراف معیار، مناسب است.
- متنی (Character): رشتههای متنی، مانند name <- c(“Adam”, “Ben”) یا شناسههای ژن (مانند “BRCA1”). این نوع برای ذخیره اطلاعات توصیفی یا برچسبها استفاده میشود.
- منطقی (Logical): مقادیر TRUE یا FALSE، مانند is_adult <- age >= 18 که برای فیلتر کردن دادهها (مثلاً شناسایی نمونههای با بیان ژن بالا) کاربرد دارد.
- فاکتور (Factor): متغیرهای دستهای که مقادیر متنی را بهصورت برچسبهای عددی کدگذاری میکنند، مانند gender <- factor(c(“male”, “female”)). فاکتورها در تحلیلهای آماری، مانند مقایسه گروههای بیماران یا دستهبندی نمونهها، بسیار مفیدند. از نسخه R 4.0.0 (آوریل 2020)، متغیرهای متنی بهطور پیشفرض به فاکتور تبدیل نمیشوند مگر اینکه صراحتاً با stringsAsFactors = TRUE مشخص شود، که این تغییر از سردرگمیهای رایج جلوگیری کرده است.
برای بررسی نوع داده، از تابع typeof() یا is.*() استفاده میشود:
typeof(age) # خروجی: “double”
is.character(name) # خروجی: TRUE
is.factor(gender) # خروجی: TRUE (اگر فاکتور باشد)
درک دقیق انواع داده به پژوهشگران زیستی کمک میکند تا از ناسازگاریها، مانند استفاده از دادههای متنی در محاسبات عددی، اجتناب کنند.
-
ساختارهای دادهای (Data Structures)
ساختارهای دادهای در R روشهایی برای سازماندهی دادهها با ترکیب شکل (Shape) و نوع داده هستند. این ساختارها به دو دسته همگن (Homogeneous) و ناهمگن (Heterogeneous) تقسیم میشوند: ساختارهای همگن فقط یک نوع داده را ذخیره میکنند، در حالی که ساختارهای ناهمگن چندین نوع داده را پشتیبانی میکنند.
-
بردارهای اتمی (Atomic Vectors)
بردارها اساسیترین ساختار دادهای در R هستند که توالی خطی از عناصر با نوع یکسان را ذخیره میکنند. ویژگیهای بردار شامل نوع (typeof())، طول (length())، و ویژگیهای اختیاری مانند نامها (names()) است. مثال:
age <- c(21, 19, 20)
length(age) # خروجی: 3
names(age) <- c(“Adam”, “Ben”, “Cecily”)
بردارها برای دادههای خطی مانند مقادیر بیان ژن، سن نمونهها، یا مقادیر p-value در تحلیلهای آماری مناسباند. محدودیت آنها این است که نمیتوانند انواع داده مختلف را بهطور همزمان ذخیره کنند.
-
لیستها (Lists)
لیستها بردارهای ناهمگنی هستند که میتوانند عناصر با انواع و اشکال مختلف (مانند اعداد، متن، بردارها، یا حتی لیستهای دیگر) را ذخیره کنند. مثال:
l <- list(1, “B”, c(3, 4), list(5, 6))
str(l) # نمایش ساختار سلسلهمراتبی
لیستها برای ذخیره دادههای پیچیده زیستی، مانند خروجیهای تحلیلهای آماری (که ممکن است شامل ضرایب، p-value، و متن توضیحی باشد)، ایدهآل هستند. دسترسی به عناصر با [[]] برای استخراج یک عنصر یا [] برای زیرلیست انجام میشود:
l[[3]] # خروجی: c(3, 4)
l[3] # خروجی: list(c(3, 4))
-
ماتریسها (Matrices)
ماتریسها بردارهای اتمی دوبعدی هستند که تمام عناصرشان باید از یک نوع باشند. با تابع matrix() ایجاد میشوند:
m <- matrix(1:6, nrow=2, ncol=3, byrow=TRUE)
ماتریسها برای دادههای همگن مانند ماتریسهای همبستگی ژنها یا ماتریسهای فاصله ژنومی در تحلیلهای فیلوژنتیک مناسباند. دسترسی به عناصر با اندیسگذاری دوبعدی انجام میشود:
m[1, 2] # خروجی: 2
-
دیتافریمها (Data Frames)
دیتافریمها ساختارهای جدولی دوبعدی هستند که ستونهایشان میتوانند انواع داده مختلف داشته باشند، اما همه ستونها باید طول یکسانی داشته باشند. مثال:
df <- data.frame(name=c(“Adam”, “Ben”), age=c(21, 19), gender=c(“male”, “male”))
دیتافریمها برای دادههای زیستی مانند اطلاعات بالینی (نام بیمار، سن، جنسیت، نوع بیماری) یا دادههای آزمایشگاهی بسیار مناسباند. دسترسی به ستونها با عملگر $ یا اندیسگذاری انجام میشود:
df$name # خروجی: c(“Adam”, “Ben”)
df[, 1] # خروجی مشابه
-
آرایهها و جداول (Arrays/Tables)
آرایهها بردارهای چندبعدی هستند که فقط یک نوع داده را ذخیره میکنند و با تابع array() ایجاد میشوند:
a <- array(letters[1:8], dim=c(2, 2, 2))
جداول (Tables) نوع خاصی از آرایهها هستند که فرکانس ترکیبهای دستهای را نشان میدهند:
table(df$gender, df$age > 20)
این ساختارها برای تحلیلهای چندبعدی، مانند بررسی فرکانس دستههای ژنوتیپ یا گروههای بیماری، کاربرد دارند، اما به دلیل پیچیدگی، کمتر از بردارها و دیتافریمها استفاده میشوند.
انتخاب ساختار دادهای مناسب
انتخاب ساختار دادهای به نوع، شکل، و هدف داده بستگی دارد:
- بردارها: برای دادههای خطی همگن، مانند مقادیر عددی (سن، بیان ژن) یا متنی (شناسه نمونه).
- لیستها: برای دادههای ناهمگن یا سلسلهمراتبی، مانند نتایج تحلیلهای آماری یا دادههای چندلایه.
- ماتریسها: برای دادههای دوبعدی همگن، مانند ماتریسهای همبستگی یا فاصله.
- دیتافریمها: برای دادههای جدولی ناهمگن، مانند دادههای بالینی یا نتایج آزمایشگاهی.
- آرایهها/جداول: برای دادههای چندبعدی یا فرکانسهای دستهای، مانند تحلیلهای پیچیده آماری.
https://bookdown.org/hneth/i2ds/struc.html
دستورات متداول در R برای مدیریت و پیشپردازش دادهها
دستورات زیر به پژوهشگران کمک میکنند تا دادههای زیستی را بهصورت کارآمد پردازش کنند.
- csv()
خواندن دادهها از فایلهای CSV، مانند دادههای بیان ژن یا اطلاعات بالینی ذخیرهشده در فرمت CSV.
- table()
وارد کردن دادهها از فایلهای متنی با ساختار جدولی، مانند فایلهای تبدلشده حاوی دادههای ژنومی.
- frame()
ایجاد یک دیتافریم از بردارها، برای سازماندهی دادههای زیستی مانند نام نمونهها، مقادیر بیان، و دستهبندیها.
- subset()
فیلتر کردن دادهها بر اساس شرایط خاص، مانند انتخاب نمونههای با بیان ژن بالای یک آستانه مشخص.
- merge()
ادغام دیتافریمهای مختلف، مانند ترکیب دادههای بیان ژن با اطلاعات متادیتای نمونهها بر اساس شناسه مشترک.
- aggregate()
جمعبندی دادهها بر اساس معیارهای خاص، مانند محاسبه میانگین بیان ژن برای هر گروه از بیماران.
- transform()
ایجاد متغیرهای جدید در دیتافریم، مانند محاسبه لگاریتم مقادیر بیان ژن برای نرمالسازی دادهها.
- sort()
مرتبسازی بردارها یا دیتافریمها، مانند مرتب کردن دادههای ژنومی بر اساس p-value یا مقادیر بیان.
- unique()
شناسایی مقادیر یکتا در یک بردار یا ستون، مانند استخراج ژنهای منحصربهفرد از یک مجموعه داده.
- ifelse()
ارزیابی شرطی برای ایجاد مقادیر جدید، مانند دستهبندی نمونهها به «بالا» یا «پایین» بر اساس سطح بیان ژن.
- apply()
اعمال یک تابع به سطرها یا ستونهای ماتریس یا دیتافریم، مانند محاسبه میانگین بیان ژن برای هر نمونه.
- lapply(), sapply(), mapply()
اعمال توابع به لیستها یا بردارها، مانند نرمالسازی مقادیر بیان ژن در چندین لیست داده.
- dplyr::filter()
فیلتر کردن دادهها در دیتافریم با استفاده از بسته dplyr، مانند انتخاب نمونههای متعلق به یک گروه بیماری خاص.
- dplyr::mutate()
ایجاد یا تغییر متغیرها در دیتافریم با بسته dplyr، مانند افزودن ستونی برای مقادیر نرمالشده بیان ژن.
- csv()
صادر کردن دادهها به فایل CSV، مانند ذخیره نتایج پیشپردازششده برای استفاده در ابزارهای دیگر.
- readRDS(), saveRDS()
خواندن و ذخیره اشیاء دادهای R، مانند ذخیره دیتافریمهای بزرگ زیستی برای استفاده مجدد سریع
https://www.ionos.com/digitalguide/websites/web-development/r-commands/
کار با بستههای تخصصی R: نصب و استفاده از Bioconductor
Bioconductor، بهعنوان یک پروژه متنباز مبتنی بر R، ابزارهای تخصصی برای مدیریت و پیشپردازش دادههای زیستی با حجم بالا، مانند دادههای NGS یا بیان ژن، ارائه میدهد. این بخش به معرفی دستورات متداول در R برای مدیریت و پیشپردازش دادهها با استفاده از بستههای Bioconductor میپردازد، با تمرکز بر ابزارهایی که بهطور خاص برای دادههای ژنومی طراحی شدهاند. این ابزارها به پژوهشگران زیستی کمک میکنند تا دادههای خام را به فرمتهای قابلتحلیل تبدیل کنند.

- BiocManager::install()
نصب بستههای اصلی Bioconductor برای آمادهسازی محیط R جهت تحلیل دادههای ژنومی، مانند دادههای RNA-Seq یا ChIP-Seq.
- library(Biobase)
بارگذاری بسته Biobase برای دسترسی به ابزارهای پایه مدیریت دادههای زیستی، مانند ساختارهای داده برای ذخیره دادههای بیان ژن و متادیتا.
- BiocManager::install(“GenomicFeatures”)
نصب بسته GenomicFeatures برای پردازش و مدیریت دادههای ژنومی، مانند استخراج اطلاعات ژنها و ترانسکریپتها از پایگاههای داده ژنومی.
- makeTxDbFromGFF()
ایجاد پایگاه داده ترانسکریپت (TranscriptDb) از فایلهای GFF/GTF، برای تحلیل دادههای ژنومی مانند حاشیهنویسی ژنها در دادههای توالییابی.
- genes()
استخراج اطلاعات ژنها از یک شیء TranscriptDb، مانند موقعیتهای ژنومی ژنها برای تحلیلهای داوناستریم.
- library(DESeq2)
بارگذاری بسته DESeq2 برای آمادهسازی دادههای بیان ژن (مانند دادههای شمارش RNA-Seq) برای تحلیل بیان تفریقی.
- DESeqDataSetFromMatrix()
ایجاد یک شیء DESeqDataSet از ماتریس شمارش ژنها و متادیتای نمونهها، برای پیشپردازش دادههای RNA-Seq قبل از تحلیل آماری.
- library(edgeR)
بارگذاری بسته edgeR برای مدیریت دادههای شمارش ژن، مانند فیلتر کردن ژنهای با بیان پایین در تحلیلهای RNA-Seq.
- DGEList()
ایجاد یک شیء DGEList از دادههای شمارش ژن، برای پیشپردازش و نرمالسازی دادههای بیان ژن در بسته edgeR.
- filterByExpr()
فیلتر کردن ژنهای با بیان پایین از یک شیء DGEList، برای بهبود کیفیت دادههای RNA-Seq قبل از تحلیل.
- library(GenomicRanges)
بارگذاری بسته GenomicRanges برای مدیریت محدودههای ژنومی، مانند همپوشانیهای ژنومی در دادههای ChIP-Seq یا ATAC-Seq.
- GRanges()
ایجاد یک شیء GRanges برای ذخیره محدودههای ژنومی (مانند کروموزوم، موقعیت شروع و پایان) و متادیتای مرتبط.
- findOverlaps()
شناسایی همپوشانی بین محدودههای ژنومی در دو شیء GRanges، مانند یافتن پیکهای ChIP-Seq که با ژنهای خاص همپوشانی دارند.
- library(SummarizedExperiment)
بارگذاری بسته SummarizedExperiment برای سازماندهی دادههای زیستی پیچیده، مانند ترکیب دادههای بیان ژن، متادیتای نمونه، و حاشیهنویسی ژنها.
- SummarizedExperiment()
ایجاد یک شیء SummarizedExperiment برای ذخیره دادههای ماتریسی (مانند شمارش ژنها) همراه با متادیتای نمونه و ویژگیها.
- assays()
دسترسی به دادههای ماتریسی (مانند شمارش ژنها) در یک شیء SummarizedExperiment، برای پیشپردازش یا استخراج دادهها.
- colData()
استخراج متادیتای نمونهها (مانند نوع نمونه یا شرایط آزمایش) از یک شیء SummarizedExperiment، برای فیلتر یا پردازش بیشتر.
- rowRanges()
دسترسی به حاشیهنویسی ویژگیها (مانند اطلاعات ژنها) در یک شیء SummarizedExperiment، برای تحلیلهای ژنومی.
https://www.geeksforgeeks.org/installation-of-bioconductor-in-r/
مقایسه R و Python در تحلیل دادههای زیستی: مزایا و معایب
R و Python دو زبان برنامهنویسی برجسته در تحلیل دادههای زیستی، مانند دادههای NGS، بیان ژن، و تحلیلهای پروتئومیکس، هستند که هرکدام ویژگیهای منحصربهفردی برای پژوهشهای زیستی ارائه میدهند. این بخش به بررسی مزایا و معایب این دو زبان در زمینه زیستفناوری تمرکز دارد، با تأکید بر کاربردهای خاص زیستی مانند مدیریت دادههای ژنومی، حاشیهنویسی ژنها، و یکپارچهسازی دادههای چندلایه.
مزایای R در تحلیل دادههای زیستی
- طراحی تخصصی برای تحلیل آماری زیستی:
R با هدف تحلیل آماری و گرافیکی طراحی شده است و بستههایی مانند Bioconductor (با بیش از 2000 بسته تخصصی) بهطور خاص برای دادههای ژنومی، مانند تحلیل بیان تفریقی (DESeq2) یا حاشیهنویسی ژن (GenomicFeatures)، ارائه میدهد. این تمرکز R را برای پژوهشگران زیستی که به ابزارهای آماری آماده نیاز دارند، ایدهآل میکند.
- بصریسازی پیشرفته با قابلیتهای زیستی:
R با بستههایی مانند ggplot2 و ComplexHeatmap امکان ایجاد نمودارهای پیچیده زیستی، مانند نقشههای حرارتی برای دادههای ژنومی یا نمودارهای آتشفشانی برای بیان ژن، را با کیفیت بالا و انعطافپذیری زیاد فراهم میکند، که در زیستفناوری برای ارائه نتایج بصری دقیق حیاتی است.
- جامعه کاربری قوی در بیوانفورماتیک:
جامعه R، بهویژه از طریق CRAN و Bioconductor، بهطور فعال در توسعه ابزارهای زیستی مشارکت دارد. این جامعه استانداردهای سختگیرانهای برای مستندسازی (مانند vignettes) ارائه میدهد، که یادگیری و کاربرد ابزارهای زیستی مانند تحلیل RNA-Seq را برای پژوهشگران آسانتر میکند.
معایب R در تحلیل دادههای زیستی
- منحنی یادگیری پیچیده برای وظایف پیشرفته:
گرچه R برای تحلیلهای پایه زیستی ساده است، وظایف پیچیدهتر، مانند یکپارچهسازی دادههای چندمنبع (مانند پروتئومیکس و ژنومیکس)، به دلیل سینتکس غیرشهودی و تنوع بستهها میتواند چالشبرانگیز باشد، بهویژه برای پژوهشگران با تجربه برنامهنویسی محدود.
- عملکرد محدود در دادههای بسیار بزرگ:
R در مقایسه با Python برای مدیریت دادههای زیستی با حجم بسیار بالا (مانند دادههای توالییابی کلژنوم) کندتر است، زیرا بهینهسازیهای کمتری برای پردازش موازی یا مدیریت حافظه دارد، که در پروژههای مقیاس بزرگ زیستی میتواند محدودیت ایجاد کند.
مزایای Python در تحلیل دادههای زیستی
- تطبیقپذیری در پروژههای چندرشتهای زیستی:
Python بهعنوان یک زبان همهمنظوره، امکان یکپارچهسازی تحلیل دادههای زیستی با سایر حوزهها، مانند یادگیری ماشین (با TensorFlow) یا توسعه ابزارهای وب زیستی (با Django)، را فراهم میکند. این ویژگی برای پروژههای زیستی چندرشتهای، مانند توسعه مدلهای پیشبینی بیماری، بسیار مفید است.
- عملکرد بالا در دادههای بزرگ زیستی:
Python با کتابخانههایی مانند NumPy و Pandas برای مدیریت دادههای ژنومی بزرگ (مانند فایلهای VCF یا BAM) بهینهتر است و از پردازش موازی با ابزارهایی مانند Dask پشتیبانی میکند، که برای تحلیل دادههای توالییابی با حجم بالا حیاتی است.
- یادگیری آسانتر برای مبتدیان زیستی:
سینتکس ساده و مشابه زبان طبیعی Python یادگیری آن را برای پژوهشگران زیستی بدون تجربه برنامهنویسی آسانتر میکند. کتابخانههای زیستی مانند Biopython نیز ابزارهای سادهای برای وظایف مانند تجزیه فایلهای FASTA یا BLAST ارائه میدهند.
معایب Python در تحلیل دادههای زیستی
- اکوسیستم زیستی کمتر تخصصی:
در مقایسه با Bioconductor در R، اکوسیستم زیستی Python (مانند Biopython یا Pybedtools) کمتر جامع است و ابزارهای کمتری برای تحلیلهای آماری زیستی تخصصی، مانند بیان تفریقی یا حاشیهنویسی ژنومی، ارائه میدهد.
- وابستگی به کتابخانههای خارجی:
Python برای تحلیلهای زیستی پیچیده اغلب به کتابخانههای متعدد (مانند Pandas، SciPy، و Biopython) وابسته است، که میتواند مدیریت وابستگیها و سازگاری نسخهها را برای پژوهشگران زیستی که تجربه برنامهنویسی محدودی دارند، دشوار کند.
در نهایت، انتخاب بین R و Python در تحلیل دادههای زیستی به نیازهای پروژه و پیشزمینه پژوهشگر بستگی دارد. R برای تحلیلهای آماری زیستی تخصصی و بصریسازی با کیفیت بالا برتری دارد، در حالی که Python برای پروژههای مقیاس بزرگ، چندرشتهای، و یادگیری آسانتر مناسبتر است. استفاده ترکیبی از هر دو زبان، مثلاً پیشپردازش دادهها با Python و تحلیل آماری با R، میتواند بهترین نتیجه را در پژوهشهای زیستی به همراه داشته باشد.
بصری سازی دادهها در R با استفاده از ggplot2 و Plotly
بسته ggplot2 در زبان R یکی از قدرتمندترین ابزارها برای بصری سازی دادههای زیستی، از جمله دادههای ژنومی مانند NGS، است که بر اساس مفهوم «گرامر گرافیک» (Grammar of Graphics) طراحی شده است.
این مفهوم به این معناست که یک نمودار از ترکیب لایههای مختلف، مانند دادهها، اشکال هندسی، مقیاسها و توضیحات، ساخته میشود. برخلاف سیستم گرافیکی پایه R که بهصورت مستقیم نمودارها را ترسیم میکند، ggplot2 به کاربران اجازه میدهد تا با افزودن لایههای مختلف، نمودارهای پیچیده و حرفهای را بهصورت گامبهگام ایجاد کنند.
این انعطافپذیری، ggplot2 را برای نمایش دادههای زیستی، مانند بیان ژنها یا توزیع توالیها، بسیار مناسب میکند. به زبان ساده، ggplot2 مانند یک بوم نقاشی است که پژوهشگر میتواند با ابزارهای مختلف، تصویر دلخواه از دادهها را خلق کند.
همچنین بخوانید:
منابع:
https://learn.gencore.bio.nyu.edu/pre-requisites/introduction-to-linux/
https://omicstutorials.com/comprehensive-guide-to-setting-up-and-using-linux-for-bioinformatics-analysis/
https://dev.to/softwaresennin/linux-directory-structure-simplified-a-comprehensive-guide-3012
https://www.geeksforgeeks.org/introduction-linux-shell-shell-scripting/
http://www.sixthresearcher.com/list-of-helpful-linux-commands-to-process-fastq-files-from-ngs-experiments/
https://library.fiveable.me/biostatistics/unit-13/introduction-rstudio-biological-data-analysis/study-guide/hBDIMXTbKGr2Js6p
https://www.geeksforgeeks.org/how-to-install-r-studio-on-windows-and-linux/https://rstudio-education.github.io/hopr/starting.html