



سندرم X شکننده چیست؟
سندرم X شکننده با ناتوانی ذهنی متوسط در مردان مبتلا و ناتوانی ذهنی خفیف در زنان مبتلا مشخص میشود. گاهی اوقات ویژگیهای فیزیکی متمایز در مردان مبتلا وجود دارد که شامل سر بزرگ، صورت دراز، پیشانی و چانه برجسته، گوشهای بیرون زده، مفاصل شل و بیضههای بزرگ است اما این ویژگیها با گذشت زمان ایجاد میشوند و ممکن است تا سن بلوغ آشکار نشوند. تأخیرهای حرکتی و زبانی معمولاً وجود دارند اما به مرور زمان آشکارتر میشوند. ناهنجاریهای رفتاری از جمله رفتارهای اوتیستیک (autistic behaviors) شایع هستند.
سندرم X شکننده در تمام اقوام و نژادهای اصلی یافت شده است و ناشی از یک ناهنجاری (جهش) در ژن FMR1 است. FMR1 ژنی است که بر روی کروموزوم X قرار دارد و پروتئینی به نام FMRP تولید میکند که برای عملکرد مناسب سلول مورد نیاز است. این سندرم به عنوان سندرم X شکننده شناخته شد زیرا برخی از افراد مبتلا به این اختلال دارای بخشی از کروموزوم X خود بودند که به نظر میرسید شکسته یا شکننده است (اگرچه کاملاً قطع نشده است). بعداً مشخص شد که ژن FMR1 دقیقاً در جایی قرار دارد که کروموزوم X در افراد مبتلا “شکننده” به نظر میرسد.
کروموزومهایی که در هسته سلولهای انسانی وجود دارند، حامل اطلاعات ژنتیکی هر فرد هستند. سلولهای بدن انسان به طور معمول دارای 46 کروموزوم هستند. جفت کروموزومهای انسانی از 1 تا 22 شماره گذاری میشوند و کروموزومهای جنسی X و Y مشخص میشوند. مردان دارای یک کروموزوم X و Y و مادهها دارای دو کروموزوم X هستند.
هر کروموزوم دارای یک بازوی کوتاه به نام “p” و یک بازوی بلند با نام “q” است. کروموزومها بیشتر به نوارهای متعددی تقسیم میشوند که شماره گذاری میشوند. به عنوان مثال، “کروموزوم Xq27.3” به نوار 27.3 در بازوی بلند کروموزوم X که در آن ژن FMR1 قرار دارد، اشاره دارد. نوارهای شماره گذاری شده محل هزاران ژن موجود در هر کروموزوم را مشخص میکند.
اختلالات غالب مرتبط با X مانند سندرم X شکننده توسط یک ژن غیر طبیعی واقع در کروموزوم X ایجاد میشود. زنان دارای ژن غیر طبیعی ممکن است تحت تأثیر این اختلال قرار گیرند. مردان معمولاً شدیدتر از زنان مبتلا میشوند.
این فقدان یا کاهش شدید پروتئین ساخته شده توسط ژن FMR1، FMRP است که باعث ایجاد سندرم X شکننده میشود. جهش ژن FMR1 باعث از بین رفتن یا کاهش FMRP میشود. تقریباً همه افراد مبتلا دارای ناپایداری در ژن هستند که منجر به افزایش تعداد کپیهای بخشی از ژن به نام ناحیه تکرار CGG میشود (که گاهی اوقات به آن منطقه تکرار «تری نوکلئوتید» یا «سهگانه یا triplet» نیز میگویند).
هنگامی که بیش از 200 تکرار وجود داشته باشد، تغییرات شیمیایی غیر طبیعی در FMR1 رخ میدهد که متیلاسیون (methylation) نامیده میشود. گسترش ناحیه تکرار CGG به بیش از 200 تکرار همراه با متیلاسیون ژن، به نام “جهش کامل”، باعث از بین رفتن FMRP میشود که منجر به سندرم X شکننده میشود. سندرم X شکننده بیشتر در مردان رخ میدهد و منجر به اختلال شدیدتر در مردان میشود.
جهش در FMR1 در مقایسه با جهشهای موجود در ژنهای دیگر غیر معمول است. برخی از افراد بین 55 تا 200 تکرار CGG به نام “پیش موتاسیون یا premutation” را دارند، معمولاً بدون داشتن علائم مرتبط با سندرم X شکننده. این افراد در معرض خطر داشتن فرزندان یا نوههایی با سندرم X شکننده هستند اما در خطر ابتلا به دو اختلال شروع بزرگسالان، سندرم X لرزش-آتاکسی شکننده (fragile X tremor-ataxia syndrome یا FXTAS) و نارسایی اولیه تخمدان (primary ovarian insufficiency یا POI) هستند. این شرایط را اختلالات مرتبط با FMR1 نامیده اند.
مترادفها
- محل شکننده یا fragile site، نوع اسید فولیک نادر، Fra(X) (Q27.3)
- سندرم X نشانگر (marker X syndrome)
- سندرم مارتین بل (Martin-Bell syndrome)
علائم و نشانهها
سندرم X شکننده با ناتوانی ذهنی متوسط در مردان مبتلا و ناتوانی ذهنی خفیف در زنان مبتلا مشخص میشود. ویژگیهای فیزیکی در مردان مبتلا متغیر است و ممکن است تا زمان بلوغ آشکار نباشد. این علائم میتواند شامل سر بزرگ، صورت دراز، پیشانی و چانه برجسته، گوشهای بیرون زده، مفاصل شل و بیضههای بزرگ باشد.
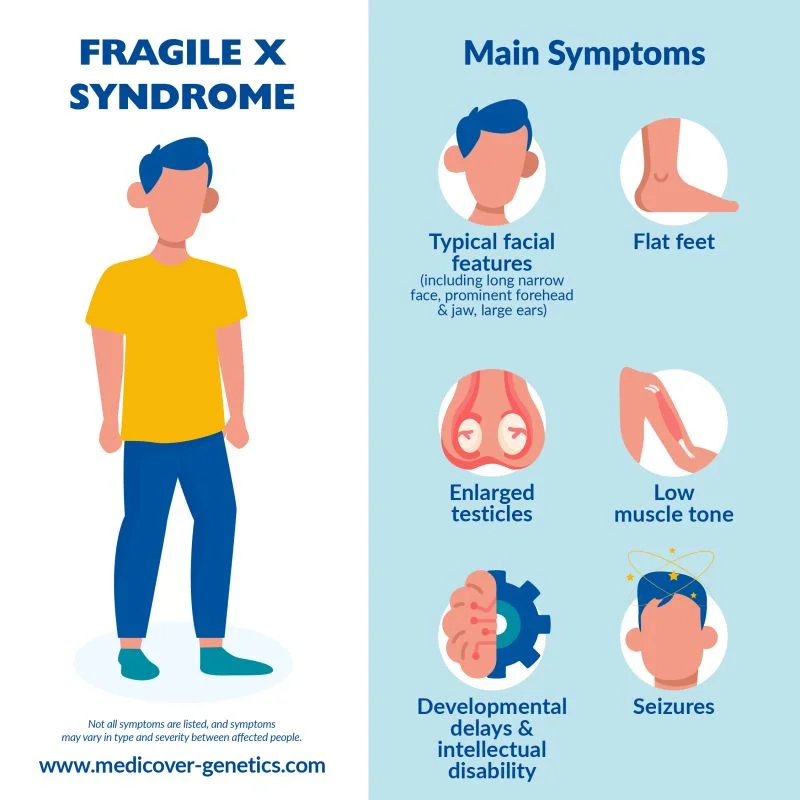
سایر علائم میتواند شامل صافی کف پا، عفونتهای مکرر گوش، تون عضلانی کم، صورت بلند و باریک، کام بالا، مشکلات دندانی، چشمهای ضربدری (استرابیسم یا strabismus) و مشکلات قلبی از جمله افتادگی دریچه میترال (mitral valve prolapse) باشد. تاخیر در رشد حرکتی، بیش فعالی، مشکلات رفتاری، راه رفتن با انگشتان پا و یا تشنجهای گاه به گاه نیز میتواند در برخی از بیماران رخ دهد. رفتارهای اوتیستیک مانند تماس چشمی ضعیف، تکان دادن دست و یا رفتارهای خود تحریکی نیز رایج هستند. تاخیرهای حرکتی و زبانی معمولاً وجود دارد اما با گذشت زمان آشکارتر میشود.
علل
همان طور که در بالا ذکر شد، سندرم X شکننده به دلیل جهش در ژن FMR1 واقع در کروموزوم X در Xq27.3 ایجاد میشود. افراد مبتلا به سندرم X شکننده تقریباً همیشه (در بیش از 99 درصد موارد) یک جهش کامل در ژن FMR1 دارند که به این معنی است که آنها بیش از 200 تکرار CGG و متیلاسیون غیر طبیعی ژن دارند.
متیلاسیون یک تغییر شیمیایی در DNA است که حامل کد ژنتیکی یک ژن است و متیلاسیون غیر طبیعی مرتبط با سندرم X شکننده باعث میشود که ژن قادر به تولید FMRP، پروتئین ساخته شده توسط ژن FMR1 که برای رشد طبیعی لازم است، نباشد. در موارد نادر، برخی از بیماران مبتلا به سندرم X شکننده به دلیل حذف DNA در کروموزوم X که در آن FMR1 قرار دارد، ژن FMR1 را تا حدی یا به طور کامل از دست میدهند و به این سندرم مبتلا هستند زیرا سلولهای آنها FMRP تولید نمیکنند.
بیماران بسیار نادر مبتلا به سندرم X شکننده، دارای جهش در یک پایه DNA هستند (موسوم به جهش نقطه ای یا point mutations) که منجر به عدم وجود یا نقص FMRP میشود. FMRP در ایجاد ارتباط بین نورونها (سلولهای عصبی) در مغز نقش دارد. فقدان یا کاهش شدید این پروتئین منجر به علائم سندرم X شکننده میشود.
پیش موتاسیونها 55-200 تکرار CGG دارند و به طور بالقوه ناپایدار هستند. افرادی که دارای پیش موتاسیون هستند، سندرم X شکننده را ندارند اما در معرض خطر ابتلا به اختلالات FMR1 FXTAS و POI در بزرگسالی هستند.
هنگامی که از نسلی به نسل دیگر منتقل میشود، ممکن است پیشجهش ناپایدار باشد و به جهش کامل تبدیل شود اما خطر بیثباتی بسته به اینکه حامل پیش جهش زن یا مرد در حال انتقال پیش جهش باشد، متفاوت است. زنانی که دارای پیش جهش ژن FMR1 هستند در معرض خطر داشتن فرزندانی با سندرم X شکننده هستند زیرا تعداد تکرارهای CGG میتواند با انتقال ژن به نسل بعدی افزایش یابد. هرچه تعداد کپیهای CGG در یک پیشجهش بیشتر باشد، احتمال اینکه این جهشها به یک جهش کامل تبدیل شوند و باعث سندرم X شکننده در فرزندان شود، بیشتر میشود.
هنگامی که نرهای دارای پیش جهش تولید مثل میکنند، فرزندان مذکر آنها هیچ خطری برای به ارث بردن پیش جهش ندارند زیرا پدران یک کروموزوم X را به پسران خود نمیدهند.
در مقابل، فرزندان دختری که پدرانشان پیش جهش دارند همیشه آن را به ارث میبرند و بنابراین نوههای پسر مبتلا به پیشجهش در خطر ابتلا به سندرم X شکننده هستند. از آن جایی که پیشجهش زمانی که از پدر به دختر منتقل میشود، نسبتاً پایدار است، دختران تقریباً هرگز به سندرم X شکننده مبتلا نمیشوند. با این حال، فرزندان آنها در معرض خطر بیشتری قرار دارند، زیرا ممکن است پیش جهش زمانی که به نسل بعدی منتقل شود ناپایدار باشد.
ژنهای طبیعی FMR1 تقریباً 5-44 تکرار CGG دارند و این تعداد از نسلی به نسل دیگر ثابت میماند. گاهی اوقات، در برخی از افراد با 45-54 تکرار، ناپایداری جزئی وجود دارد به طوری که این افراد چندین بار بیشتر (یا کمتر) از والدین خود تکرار میکنند. یک عدد تکرار FMR1 بین 45 و 54 “متوسط” یا “منطقه خاکستری یا gray zone” نامیده میشود اما این ناپایداری جزئی منجر به هیچ نشانه ای از سندرم X شکننده یا اختلالات مربوط به FMR1 نمیشود. داشتن تعداد متوسطی از تکرارهای CGG همچنان در محدوده نرمال تعداد تکرار در نظر گرفته میشود.
جمعیتهای آسیب دیده
سندرم X شکننده از هر 4000 مرد 1 نفر و در ایالات متحده آمریکا از هر 6000 تا 8000 زن 1 نفر را تحت تاثیر قرار میدهد. یعنی تقریباً دو برابر بیشتر از زنان در مردان تأثیر میگذارد. با این حال، به نظر میرسد حدود چهار برابر تعداد زنها ناقل ژن تغییر یافته نسبت به مردان (1:250 زن و 1:1000 مرد) است. سندرم X شکننده در تمام اقوام و نژادهای اصلی یافت شده است.
اختلالات با علائم مشابه
پیش جهشهای ژن FMR1 با دو اختلال دیگر مرتبط است و این بیماریها را اختلالات مرتبط با FMR1 نامیده اند. همه افراد مبتلا به پیش جهش به اختلالات مرتبط با FMR1 مبتلا نمیشوند اما داشتن پیش موتاسیون خطر ابتلا به این اختلالات را افزایش میدهد.
سندرم لرزش آتاکسی X شکننده (Fragile X tremor-ataxia syndrome یا FXTAS) با ناهنجاریهای حرکتی پیشرونده در بزرگسالی (آتاکسی یا ataxia) و حرکات ریتمیک و غیرارادی (لرزش) مشخص میشود که بیشتر مردان را تحت تأثیر قرار میدهد. افراد مبتلا به این بیماری دارای یک پیش موتاسیون در ژن FMR1 هستند (55-200 تکرار CGG). تشخيص FXTAS را مي توان با شباهت آن به ساير اختلالات ديررس بزرگسالان مانند بيماري پاركينسون پيچيده كرد.
نارسایی اولیه تخمدان (POI) مربوط به FMR1 به عنوان یائسگی قبل از 40 سالگی در زنانی که دارای پیش موتاسیون در ژن FMR1 هستند (55-200 تکرار CGG) تعریف میشود. خطر POI در حاملان پیش موتاسیون تقریباً 21 درصد است. زنان مبتلا به POI با علت ناشناخته در معرض خطر 1 در50 قرار دارند تا ناقل پیش موتاسیون در ژن FMR1 باشند.
برخی از علائم اختلالات زیر میتواند مشابه علائم سندرم X شکننده باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد:
سندرم XE شکننده (Fragile XE syndrome یا FRAXE) نادر است و ناشی از یک ژن FMR2 غیر طبیعی است که در کروموزوم X بسیار نزدیک به محل ژن FMR1 قرار دارد. ژن طبیعی FMR2 حاوی 6-35 نسخه CCG است و افراد مبتلا به این اختلال بیش از 200 نسخه از CCG در ژن FMR2 دارند. اثر ژنهای FMR2 با 35-200 کپی از CCG هنوز مشخص نشده است زیرا این اختلال تظاهرات بالینی خفیفی دارد. علائم شایع FRAXE شامل ناتوانی ذهنی خفیف، نقص در یادگیری و تاخیرهای رشدی احتمالی است.
سندرم رنپنینگ (Renpenning syndrome) یکی از اختلالات ناتوانی ذهنی مرتبط با کروموزوم X است که مردان را تقریباً به استثنای زنان مبتلا میکند. به ندرت زنان با این سندرم مواجه میشوند. این بیماری با ناتوانی ذهنی مشخص میشود که میتواند شامل کوتاه قدی شدید، دور سر کوچکتر از حد طبیعی (میکروسفالی یا microcephaly) و بیضههای کوچک باشد. این سندرم به مکان نقشه ژنی Xp11.2-p11.4 نگاشت شده است و اصطلاح “سندرم رنپنینگ” باید به شرایطی محدود شود که به این منطقه اشاره میکند. شیوع این بیماری ناشناخته است.
تأخیر رشد در سندرم رنپنینگ در مراحل اولیه وجود دارد و مردان در سنین 2 تا 3 سالگی راه رفتن را یاد میگیرند و در سنین 3 تا 4 سالگی قادر به گفتن کلمات ساده هستند. اگرچه یک مرد مبتلا ممکن است از نظر فیزیکی طبیعی به نظر برسد اما دور سر و قد او در حد پایین تر از حد طبیعی است. پس از بلوغ، بیضهها کوچکتر از حد طبیعی خواهند بود.
تشخیص بیماری بسیار دشوار است، به خصوص اگر در یک خانواده فقط یک مرد با ناتوانی ذهنی وجود داشته باشد. تشخیص باید بر اساس شواهدی مبنی بر وراثت به عنوان یک صفت مرتبط با X و تعیین اینکه ژن آسیب دیده بر روی بازوی کوتاه (در Xp11.1-p11.4) کروموزوم X قرار دارد، باشد.
تشخیص
بیش از 99 درصد از افراد مبتلا به سندرم X شکننده دارای جهش کامل (بیش از 200 تکرار CGG و متیلاسیون غیر طبیعی) در ژن FMR1 هستند. آزمایش ژنتیک مولکولی برای تعیین تعداد تکرارهای CGG در ژن FMR1 و آزمایش برای تعیین وضعیت متیلاسیون ژن FMR1 اغلب برای پیگیری یافتن یک ناحیه CGG گسترش یافته استفاده میشود.
تجزیه و تحلیل کروموزوم با استفاده از تکنیکهای ویژه برای القای مکانهای شکننده در کروموزومها زمانی برای تشخیص سندرم X شکننده استفاده میشد اما دیگر برای این منظور استفاده نمیشود. سندرم X شکننده نامی است که به این بیماری داده شده است زیرا برخی از افراد مبتلا یک کروموزوم X دارند که به نظر میرسد “شکسته” یا “شکننده” است و با کوچکترین پیوندی به هم چسبیده است. این روش دیگر در تشخیص این سندرم استفاده نمیشود زیرا هم دقت کمتری دارد و هم هزینه بیشتری نسبت به تکنیکهای مولکولی دارد.
درمان
درمانهای زیادی برای سندرم X شکننده وجود دارد که میتواند زندگی افراد مبتلا و خانوادههای آنها را بهبود بخشد. اینها شامل آموزش ویژه، گفتار، آموزش شغلی و یکپارچگی حسی و برنامههای اصلاح رفتار است. با تلاشهای آموزشی، درمانی و حمایتی، همه افراد مبتلا به سندرم X شکننده میتوانند پیشرفت کنند. سایر درمانها ممکن است به علائم خاص فرد مبتلا بستگی داشته باشد. مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود.
تعداد زیادی کلینیک Fragile X در ایالات متحده و در سراسر جهان وجود دارد. این کلینیکها در درمانها، درمانها و حمایت از افراد مبتلا به سندرم X شکننده تخصص دارند و میتوانند والدین را به گزینههای دارویی برای رفع علائم خاص راهنمایی کنند. احتمالاً داروهای جدیدی برای درمان افراد مبتلا در دسترس خواهد بود و کلینیکهای تخصصی میتوانند به والدین با اطلاعات فعلی کمک کنند.
آزمایشات و مطالعات بالینی
توسعه مدلهای حیوانی برای مطالعات سندروم X شکننده در دو دهه گذشته منجر به پیش بینی زیادی برای کشف درمانهای دارویی موثر شده است. برخی از داروهایی که در ابتدا برای درمان سایر اختلالات ساخته شده بودند، در درمان علائم سندرم X شکننده در مدلهای حیوانی مؤثر بودند اما زمانی که بر روی افراد انسانی استفاده میشد، به نظر میرسید که تأثیر کمتری داشته باشند.
با این حال، محققان زیادی وجود دارند که به طور فعال روی درمانهای سندرم X شکننده کار میکنند. FRAXA، سازمانی که فعالانه در جستجوی درمان سندرم X شکننده است، بودجه جمع آوری کرده و از تلاشهای گسترده برای تحقیقات X شکننده حمایت میکند. وب سایت FRAXA (www.fraxa.org) یک منبع عالی از اطلاعات جاری در مورد پروژههای تحقیقاتی و آزمایشات بالینی آنها است.
همچنین بخوانید:
مترجم: فاطمه فریادرس