



نام های دیگر سندرم کری دو چت
- سندرم p5
- سندرم فریاد گربه
- CDCS
- سندرم لژون (Lejeune syndrome)
مباحث عمومی
سندرم کری دو چت (CdCS or 5p) یک اختلال ژنتیکی نادر است که در آن بخش متغیری از بازوی کوتاه کروموزوم 5 از بین رفته یا حذف شده است (monosomic، مونوسومی). بسته به اندازه و محل دقیق ماده ژنتیکی حذف شده، علائم در هر مورد بسیار متفاوت هستند.
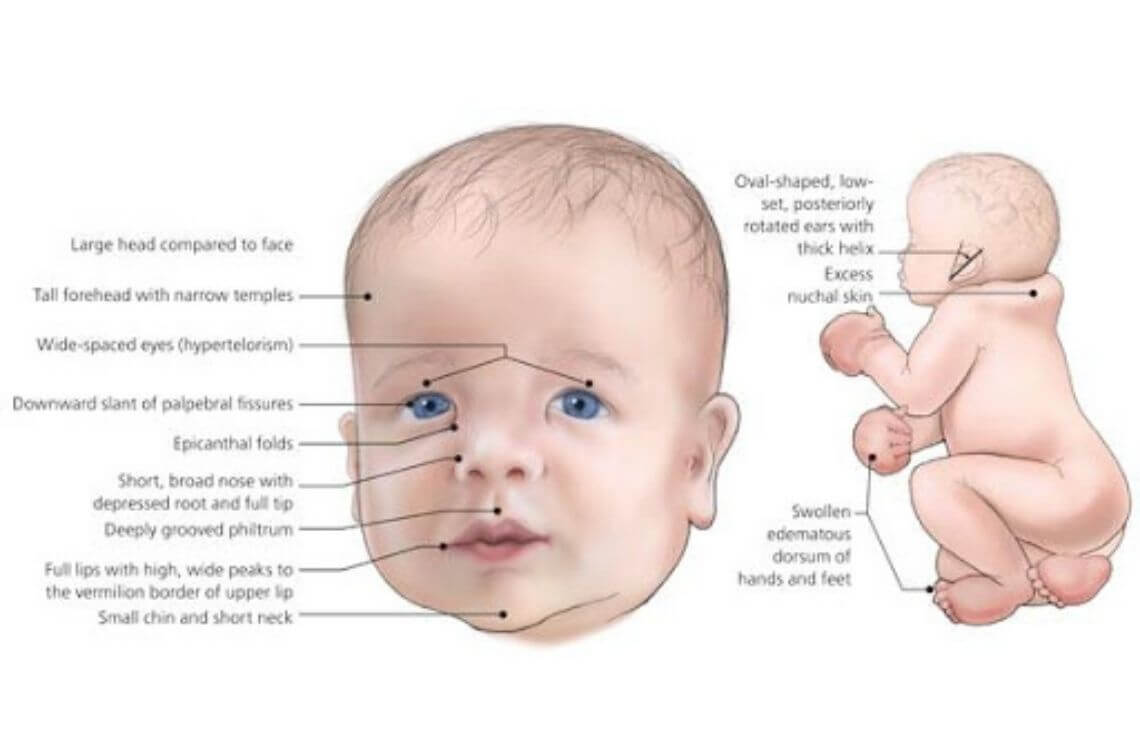
علائم متداول عبارتند از: گریه متمایز از سایر کودکان و شبیه به صدای گربه، ویژگیهای شاخص در صورت، رشد آهسته و میکروسفالی (microcephaly)، وضعیتی که نشان دهنده دور سر کوچکتر از حد انتظار با توجه به سن و جنس نوزاد است.
کودکان مبتلا همچنین در کسب مهارتهایی که نیاز به هماهنگی سیستمهای عضلانی و ذهنی دارند (نا توانی روانی حرکتی)، تاخیر نشان داده و ناتوانی ذهنی متوسط تا شدید را دارا هستند.
علائم اضافی دیگر که بر سیستمهای مختلف و اندامهای بدن تأثیر میگذارد نیز میتواند رخ دهد. تصور میشود که اکثر موارد ابتلا به این سندروم از خطاهای ژنتیکی خود به خودی ( de novo) در مراحل اولیه در رشد جنینی ناشی میشوند.
معرفی
این اختلال اولین بار در ادبیات پزشکی در سال 1963 توسط دکتر Lejeune توصیف شد که نام این اختلال را برگرفته از شاخصه گریه متمایز و گربه مانند آن نامید. در فرانسوی،Cri du chat به “گریه گربه” ترجمه میشود.
علائم و نشانههای سندرم فریاد گربه
علائم سندرم کری دو چت در هر مورد مبتلا متفاوت است. فریاد بلند و تیز مشخصه مرتبط با سندرم کری دو چت در چند هفته اول زندگی است. این گریه که شبیه صدای میو میو کردن گربه است، با بزرگتر شدن و افزایش سن نوزادان مبتلا کمتر میشود.

نوزادان مبتلا همچنین ممکن است وزن کم در هنگام تولد، کمبود رشد، کاهش توده عضلانی (هیپوتونی) و میکروسفالی را نشان دهند، وضعیتی که نشان دهنده دور سر کوچکتر از حد انتظار با توجه به سن و جنس نوزاد است.
ویژگیهای متمایز کننده صورت ممکن است شامل صورتهای غیرطبیعی گرد یا چاق (ماه مانند)، پل بینی پهن، چشمهای با فاصله زیاد (هیپرتلوریسم)، انحراف چشم (استرابیسم)، چینهای پلک مایل رو به پایین (palpebral fissures)، چینهای پوستی عمودی که ممکن است چشمها را در گوشههای داخلی (چین های اپیکانتال) بپوشاند، گوشهای کم شنوا و یک فک غیر طبیعی کوچک (میکروگناتیا) باشد. تراز نامناسب دندانهای بالا و پایین (مال اکلوژن) نیز ممکن است در این افراد رخ دهد.
مشخصههای دیگر صورت در این مبتلایان شامل فاصله غیر طبیعی از لب بالایی تا بینی (فیلتروم کوتاه)، بسته شدن ناقص سقف دهان (شکاف کام)، شیار یا شکاف غیر طبیعی در لب بالایی (شکاف لب) است. علاوه بر این، توده گوشتی (uvula) که در پشت گلو آویزان است ممکن است تخریب شود (bifid uvula). با بالا رفتن سن نوزادان مبتلا، صورت ممکن است چاق و پر بودن خود را از دست داده و به طور غیر طبیعی دراز و باریک شود.
بیشتر نوزادان مبتلا نیز درجاتی از ناتوانی روانی حرکتی و ذهنی را نشان میدهند. ناتوانی روانی حرکتی تاخیر در کسب مهارتهایی است که نیاز به فعالیتهای ذهنی و عضلانی مانند کنترل سر، نشستن و راه رفتن دارند. حدود نیمی از کودکان مبتلا به سندرم کری دو چت تا سن 5 سالگی توانستند لباس بپوشند.
ناتوانی ذهنی متوسط تا شدید در بیشتر موارد مبتلا وجود دارد. رشد گفتاری به ویژه در کودکان مبتلا به سندرم کری دو چت به تاخیر میافتد. کودکان مبتلا معمولاً گفتوگوها را بهتر از ارتباط برقرار کردن با دیگران درک میکنند. برخی از کودکان ممکن است بیش فعالی یا رفتارهای مرتبط با خود آزاری از خود نشان دهند. در حالی که کودکان مبتلا به سندرم کری دو چت، با هیپوتونیک (توان عضلانی کم) به دنیا میآیند اما با افزایش سن، به هیپرتونیک (توان عضلانی بالا) متمایل میشوند.
شیرخواران مبتلا ممکن است به دلیل توان عضلانی کم، مکیدن ضعیف و بیماری ریفلاکس معده دچار مشکلات تغذیهای شوند. برخی نیز در معرض خطر آسپیراسیون (aspiration) هستند که میتواند منجر به ابتلا به ذاتالریه شود. در یک مطالعه، تنها 50 درصد از کودکان مبتلا به سندرم کری دو چت تا 3.5 سالگی توانستند با قاشق غذا بخورند.
انواع یافتههای دیگر ممکن است در ارتباط با سندرم کری دو چت به دست آید. انحنای غیر طبیعی ستون فقرات از پهلو به پهلو (اسکولیوز) یک عارضه شایع در این مبتلایان است. کودکان مبتلا همچنین در معرض خطر بالاتری برای ابتلا به عفونت گوش و کاهش شنوایی هستند. تقریباً 15 تا 20 درصد از نوزادان، مبتلا به نقایص مادرزادی قلبی هستند. شایعترین نقص قلبی مجرای شریانی باز است، وضعیتی که در آن گذرگاه (مجرای) بین رگ خونی که به ریهها (شریان ریوی) و شریان اصلی بدن (آئورت) منتهی میگردد، پس از تولد بسته نمیشود.
یافتههای کمتر رایج مرتبط با سندرم کری دو چت شامل ایجاد پارگی در بافت پایهای قسمت تحتانی شکم (فتق اینگوینال) است که باعث میشود تا بخشی از روده از شکم بیرون بزند. عبور یا برگشت (ریفلاکس) محتویات معده یا روده کوچک (دئودنوم) به مری (رفلاکس معده به مری)، ناهنجاریهای کلیه و مجاری ادراری؛ مشکلات تنفسی؛ نرم شدن انگشتان دست و پا (سینداکتیلی)؛ خم شدن یا انحنای غیر طبیعی رنگ صورتی به سمت داخل انگشت چهارم (کلینوداکتیلی)؛ پای پرانتزی و ناهنجاریهای ساختاری جعبه صوتی (حنجره) و در برخی موارد، نزدیک بینی (myopia) و آب مروارید هم ممکن است ایجاد شود.
در این مبتلایان سفیدی زودرس موها نیز گزارش شده است. برخی از افراد ممکن است دچار عفونتهای تنفسی و رودهای مکرر شوند. در نوزادان پسر مبتلا، بیضهها ممکن است نتوانند به داخل کیسه بیضه پایین بیایند (کریپتورکیدیسم) و دهانه ادرار ممکن است در قسمت زیرین آلت تناسلی (هیپوسپادیاس) قرار گیرد. همچنین ثابت شده که ارتباطی بین سندروم کری دو چت و بیماری هیرشپرونگ (Hirschsprung’s disease) وجود دارد.
علل
سندرم کری دو چت یک اختلال کروموزومی است که به دلیل حذف جزئی (مونوسومی) و با طول متفاوت دز بازوی کوتاه (p) کروموزوم 5 ایجاد میشود. کروموزومهایی که در هسته سلولهای انسانی وجود دارند، حامل اطلاعات ژنتیکی برای هر فرد هستند. جفت کروموزومهای انسان از 1 تا 22 شماره گذاری میشوند و جفت 23 کروموزوم جنسی اضافی شامل یک کروموزوم X و Y در مردان و دو کروموزوم X در زنان است. هر کروموزوم دارای یک بازوی کوتاه به نام “p” و یک بازوی بلند با نام “q” است.
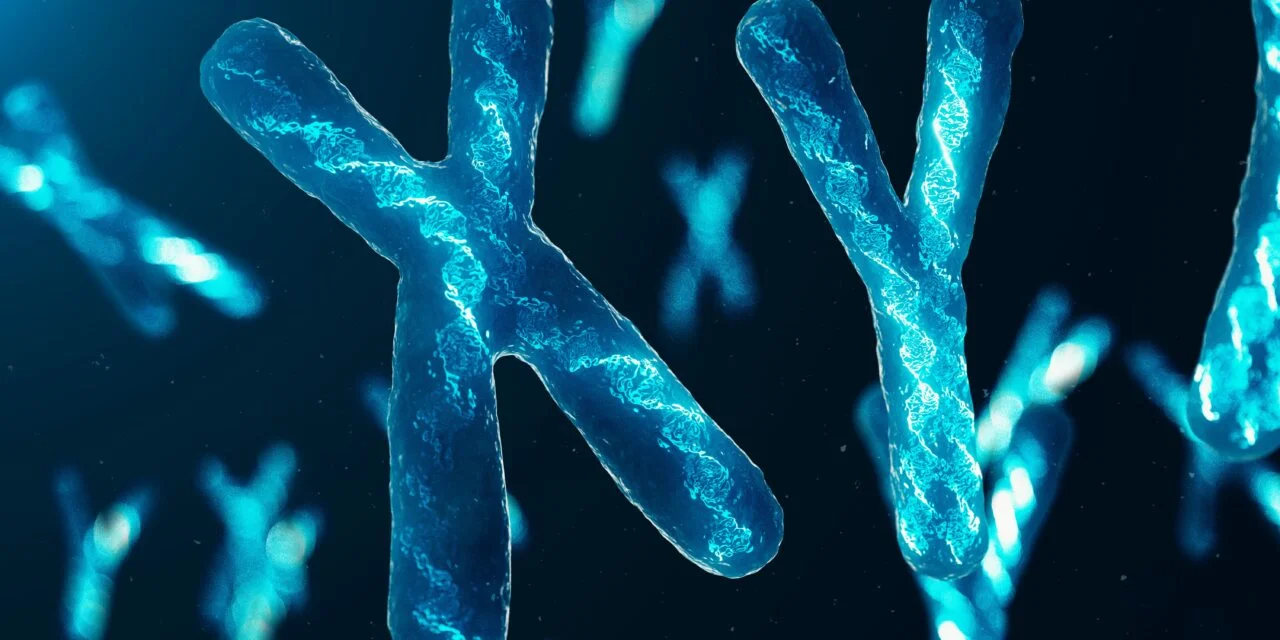
اغلب کروموزومها به قسمتهای متعددی تقسیم شده و شمارهگذاری میشوند. به عنوان مثال، “کروموزوم p15.35” به قسمت 15 در بازوی کوتاه کروموزوم 5 اشاره دارد. بخشهای شماره گذاری شده محل هزاران ژن موجود در هر کروموزوم را مشخص میکنند.
در افراد مبتلا به سندرم کری دو چت، دامنه و شدت علائم و مشخصههای مرتبط افراد میتواند متفاوت باشد (بسته به طول یا محل دقیق قسمت حذف شده کروموزوم p5). محققان تشخیص دادهاند که وجود علائم خاصی ممکن است با مناطق خاصی در بازوی کوتاه کروموزوم 5 مرتبط باشد.
محققان چندین ژن را شناسایی کردهاند که اعتقاد بر این است که در ایجاد سندرم کری دو چت نقش دارند. ژن ترانس کریپتاز معکوس تلومراز که بر روی بازوی کوتاه کروموزوم 5 در باند 13.33 (p13.335) و ژن سمافورین F که در p15.25 قرار دارد، میتواند به تنوع گسترده ویژگیهای مربوط به این سندروم کمک کند. حذف ژن d-catenin (همچنین در p15.25) با ناتوانی ذهنی شدید تر مبتلایان مرتبط است زیرا این پروتئین در رشد اولیه عصبی بیان میشود.
اگر محققان بتوانند مجموعه خاصی از علائم و مشخصهها (فنوتیپها) را به حذف قسمت خاصی از کروموزوم p5 مرتبط کنند، ممکن است به تشخیص این بیماری و آینده بیماران ناشی از آن کمک زیادی کند.
به نظر میرسد اغلب موارد سندرم کری دو چت به دلایل ناشناخته در اوایل رشد جنینی به طور خود به خود (de novo) رخ میدهد. اکثر حذفها (۸۰ تا ۹۰ درصد) منشأ پدری دارند، به این معنی که احتمالاً به عنوان بخشی از روند تشکیل اسپرم رخ میدهند. والدین یک کودک با حذف “de novo” معمولاً کروموزومهای طبیعی داشته و میزان احتمال نسبتاً پایینی برای داشتن فرزند دیگری با ناهنجاریهای کروموزومی دارند.
تقریباً در 10 تا 15 درصد موارد، سندرم کری دو چت ممکن است ناشی از یک جابجایی متعادل (balanced translocation) بین کروموزوم p5 و کروموزوم یا کروموزومهای دیگر باشد. جابهجایی زمانی اتفاق میافتد که نواحی کروموزومهای خاصی شکسته شده و دوباره مرتب میشوند که منجر به جابجایی مواد ژنتیکی و تغییر در مجموعهای از کروموزومها میشود.
چنین جابهجایی ممکن است به دلایل نامعلوم و به شکل خود به خودی رخ دهد (de novo) یا توسط والدینی که ناقل چنین جابهجایی متعادلی هستند به فرزند (جنین) منتقل شود. جابجایی متعادل شامل مجموعهای تغییر یافته اما متعادل از کروموزومها است و معمولاً برای فرد دارای آن بی ضرر است. با این حال، چنین باز آرایی کروموزومی ممکن است با افزایش خطر رشد غیر طبیعی کروموزومی در فرزندان ناقل همراه باشد. تجزیه و تحلیل کروموزومی ممکن است تعیین کند که آیا والدین دارای جابجایی متعادل هستند یا خیر.
جمعیتهای تحت تاثیر
سندرم کری دو چت زنان را بیشتر از مردان مبتلا میکند. میزان بروز این بیماری از یک تولد در 15000 تا 50000 تولد زنده متغیر است. برخی از موارد سندرم کری دو چت ممکن است تشخیص داده نشوند و تعیین فراوانی واقعی این اختلال در جمعیت عمومی دشوار باشد.
اختلالات مرتبط
علائم اختلالات زیر میتواند شبیه به علائم سندرم کری دو چت باشد. مقایسه این اختلالات با یکدیگر ممکن است برای تشخیص افتراقی مفید باشد.
سندرم Wolf-Hirschhorn – همچنین به عنوان سندرم Wolf شناخته میشود – یک اختلال کروموزومی نادر است که در آن حذف جزئی (مونوسومی) بازوی کوتاه (p) کروموزوم 4 (p4) وجود دارد. اگر چه اندازه و محل حذف p4 از موردی به مورد دیگر متفاوت است اما اعتقاد بر این است که حذف باند p16.34 منطقه بحرانی است که منجر به ایجاد ویژگی های مشخص در این اختلال میشود.
ناهنجاریهای مرتبط معمولاً شامل وزن کم هنگام تولد، عقب ماندگی در رشد، توان عضلانی ضعیف (هیپوتونی) و تاخیر در کسب مهارتهایی است که نیاز به هماهنگی سیستمهای فیزیکی و ذهنی دارند (عقبافتادگی روانی حرکتی). اکثر نوزادان و کودکان مبتلا نیز ناهنجاریهای مشخصی در ناحیه جمجمه و صورت (psychomotor retardation) دارند.
این عارضهها ممکن است شامل سر کوچک (میکروسفالی) و پیشانی بلند باشد. ابروهای بسیار کمانی؛ چشمهایی با فاصله زیاد (هیپرتلوریسم چشمی)، چینهای پوستی عمودی که گوشههای داخلی چشم را میپوشاند (چینهای اپیکانتال)، بینی “منقاری” با پل بینی غیر طبیعی پهن، دهان رو به پایین، یک شیار عمودی کوتاه غیر معمول در وسط لب بالایی (فیلتروم)، و یا گوشهای بزرگ و بدشکل باشند.
به دلیل وجود این ناهنجاریها و یا ناهنجاریهای بخش صورت جمجمه، صورت این کودکان ممکن است از یک طرف نسبت به طرف دیگر کمی متفاوت به نظر برسد (عدم تقارن جمجمه در بخش صورت) و ناهنجاریهای فیزیکی دیگری نیز ممکن است وجود داشته باشد. چنین ویژگیهایی ممکن است شامل انحراف غیر طبیعی یک چشم نسبت به چشم دیگر باشد (استرابیسم).
به علت وجود اختلالاتی مانند عدم وجود نسبی بافت از ناحیه رنگی چشم (کلوبومای عنبیه)، بسته شدن ناقص سقف دهان (شکاف کام)، بیضههای پایین نیامده (کریپتورکیدیسم) و قرار گیری غیر طبیعی دهانه ادرار در قسمت زیرین آلت تناسلی (هیپوسپادیاس) در مردان مبتلا، ناهنجاریهای ساختاری قلب؛ دورههای ناگهانی فعالیت الکتریکی کنترل نشده در مغز (تشنج)، ناهنجاریهای اسکلتی و یا مشخصههای دیگر معمولاً به نظر میرسد که سندرم ولف-هیرشهورن (Wolf-Hirschhorn syndrome) به دلایل ناشناخته در اوایل رشد جنینی به طور خود به خود (de novo) رخ میدهد.
در موارد نادری، ممکن است این گونه به نظر برسد که این سندروم ناشی از جابجایی متعادل در یکی از والدین است.
اختلالات کروموزومی دیگری ممکن است ویژگیهای مشابه با سندرم کری دو چت داشته باشد. آزمایش کروموزومی برای تایید ناهنجاری کروموزومی خاص موجود ضروری است.
تشخیص
در نوزادان، تشخیص سندرم کری دو چت با ارزیابی بالینی کامل، شناسایی علائمهای مشخص (مثلاً گریه گربه مانند) و مطالعات کروموزومی (کاریوتایپینگ) که حذف در بازوی کوتاه کروموزوم 5 را نشان میدهد، تأیید میشود. آزمایشی که به عنوان هیبریداسیون فلورسانس در محل (FISH) شناخته میشود نیز ممکن است برای تأیید تشخیص سندرم کری دو چت استفاده شود.
همچنین ممکن است مطالعات کروموزومی برای تعیین اینکه آیا انتقال متعادل در یکی از والدین وجود دارد یا خیر انجام شود. آزمایشات تشخیصی اضافی ممکن است برای تعیین وسعت اختلال مانند اشعه ایکس برای آشکار سازی ناهنجاریهای اسکلتی مانند اسکولیوز (scoliosis) استفاده شود.
تکنیکهای علمی روز به روز در تعیین ناهنجاریهای کروموزومی اصلاح میشوند. این بدان معناست که تکنیکهای تشخیصی بهبود یافتهاند و در موارد خاص نیز تشخیص سندرم کری دو چت قبل از تولد امکان پذیر است.
درمان
درمان سندرم کری دو چت با توجه به سمت علائم خاصی است که در هر فرد آشکار است. روند درمانی ممکن است به تلاش هماهنگ تیمی از متخصصان نیاز داشته باشد.
متخصصان اطفال، ارتوپدیستها، جراحان، متخصصان قلب، آسیب شناسان گفتاری، متخصصان مغز و اعصاب، دندانپزشکان، فیزیوتراپیست و کار درمانگر و سایر متخصصان مراقبتهای پزشکی ممکن است نیاز داشته باشند که به طور سیستماتیک و جامع برای درمان کودک مبتلا برنامه ریزی کنند.
از آن جایی که برخی از کودکان مبتلا به کری دو چت ممکن است ناشنوایی حسی-عصبی داشته باشند، آزمایش شنوایی نیز باید انجام شود.
مداخله زود هنگام در حصول اطمینان از این که کودکان مبتلا به سندرم کری دو چت به بالاترین پتانسیل خود برای روند درمانی میرسند، مهم است. خدماتی که ممکن است مفید باشند ممکن است شامل آموزشهای ویژه درمانی، فیزیوتراپی، گفتار درمانی، خدمات ویژه و سایر خدمات پزشکی، اجتماعی و یا حرفهای باشند. اکثر کودکان قبل از ورود به یک سالگی در روند درمانی شرکت میکنند.
جراحی ممکن است برای درمان انواع علائم مرتبط با سندرم کری دو چت از جمله نقایص مادرزادی قلب، استرابیسم (strabismus)، اسکولیوز (scoliosis)، پای پرانتزی، شکاف کام و شکاف لب انجام شود.
میزان بقا و زنده مانی کودکان مبتلا به cri du chat به طور کلی خوب است. بیشتر مرگهای ناشی از این سندرم در سال اول زندگی کودک اتفاق میافتد. چندین فرد از این کودکان تا بالای 50 سال نیز عمر کرده اند.
مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود. درمانهای دیگر با توجه به علائم کودک و میزان پاسخ دهی وی به این روندها است.
درمانهای تحقیقاتی
تحقیقات و مطالعات سندرم کری دو چت در حال انجام است. یک مطالعه نشان داده است که مدرسههای ویژه، محیط خانه (و نه یک محیط سازمانی) و حمایت خانواده ممکن است به بیمار کمک کند تا به تواناییهای یک کودک پنج یا شش ساله عادی دست یابد. در همان مطالعه، نیمی از کودکان بالای ده سال که تحت آموزش ویژه قرار گرفته بودند و در یک محیط مرکز حمایتی زندگی میکردند، قادر به برقراری ارتباط کافی با دیگران بودند.
همچنین بخوانید:
- سندرم آشر: علائم، علل، تشخیص و درمان
- آنوریسم آئورت: بررسی اجمالی علائم، علل، درمان و پیشگیری
- سندروم مارفان: علائم، علل، تشخیص و درمان
مترجم: فاطمه فریادرس