



سندرم آپرت (Apert Syndrome) چیست؟
سندرم آپرت (Apert Syndrome) یک بیماری ژنتیکی نادر است که در بدو تولد آشکار میشود. افراد مبتلا به سندرم آپرت میتوانند ناهنجاریهای مشخصی در جمجمه، صورت، دستها و پاها داشته باشند. سندرم آپرت با کرانیوسینوستوزیس (craniosynostosis) مشخص میشود، وضعیتی که در آن مفاصل فیبری (sutures) بین استخوانهای جمجمه به شکل زودرس بسته میشوند.
این میتواند باعث شود که بالای سر نوک تیز به نظر برسد و بر استخوانهای صورت تأثیر بگذارد. برخی از انگشتان دست یا پا ممکن است به هم چسبیده یا تار مانند باشند. کودکان مبتلا ممکن است دارای ناتوانی ذهنی نیز باشند.
شدت علائم در افراد مختلف متفاوت است. سندرم آپرت تقریباً همیشه ناشی از تغییرات ژنتیکی جدید (جهش) است که به طور تصادفی رخ میدهد. به ندرت در الگوی اتوزومال غالب به ارث میرسد. افراد مبتلا به سندرم آپرت میتوانند تحت درمانهایی قرار گیرند که علائم خاصی را برطرف میکند. این میتواند شامل جراحیهای ترمیمی جمجمه، صورت، دست و پا باشد.
سندرم آپرت نوعی آکروسفالوسینداکتیلی (acrocephalosyndactyly) است. همه اشکال ACS با کرانیوسینوستوزیس مشخص میشوند و این بر رشد مناسب جمجمه و سر تأثیر میگذارد.
مترادفها
- آکروسفالوسینداکتیلی (acrocephalosyndactyly)، نوع I
- ACS1
علائم و نشانهها
سندرم آپرت با کرانیوسینوستوزیس، بسته شدن زودهنگام مفاصل فیبری (sutures) بین استخوانهای خاصی در جمجمه مشخص میشود. در افراد بدون کرانیوسینوستوز، sutures به سر نوزاد اجازه رشد و انبساط میدهند. در نهایت، این استخوانها با هم ترکیب میشوند و جمجمه را تشکیل میدهند.
برای افراد مبتلا به کرانیوسینوستوز، مغز پس از بسته شدن زودرس این sutures همچنان در حال رشد است. فشار رشد مغز میتواند باعث تغییر شکل استخوانهای مختلف جمجمه و صورت در طول رشد شود. بسته به اینکه کدام بخیه زودرس بسته میشود، شدت آن میتواند متفاوت باشد.

در بیشتر افراد مبتلا، بخیههای بین استخوانهای تشکیل دهنده پیشانی و قسمتهای بالایی جمجمه، به طور زودرس بسته میشوند. این امر باعث میشود که سر از بدو تولد نوک تیز به نظر برسد (آکروسفالی یا acrocephaly). علاوه بر این، قسمت پشتی جمجمه ممکن است صاف و با پیشانی بلند و پهن به نظر برسد.
ممکن است یک “نقطه نرم یا soft spot” بزرگ و دیر بسته روی جمجمه وجود داشته باشد. افراد همچنین ممکن است هیدروسفالی (hydrocephalus) داشته باشند که در آن مایع مغزی نخاعی به طور غیر طبیعی در حفرههای مغز تجمع مییابد. این میتواند باعث فشار بر مغز شود.
استخوانهای صورت ممکن است تحت تأثیر کرانیوسینوستوز قرار گیرند. این میتواند منجر به ناهنجاریهای مشخصه صورت شود. افراد مبتلا به سندرم آپرت ممکن است دارای فاصله زیاد چشم (هیپرتلوریسم یا hypertelorism)، چشمهای برآمده، یا شقاق کف دستی با مایل به پایین باشند.
همچنین ممکن است نواحی میانی صورت (هیپوپلازی فک بالا یا maxillary hypoplasia) و ناهنجاریهای کام مانند شکاف کام داشته باشند. سمت راست و چپ صورت ممکن است متقارن نباشند. افراد مبتلا به سندرم آپرت ممکن است بینی صاف با پل کم داشته باشند. ممکن است افراد دچار تاخیر در رشد دندان، شلوغی دندان یا اپن بایت (open bite) شوند.
آنها میتوانند آکنه متوسط تا شدید داشته باشند. اگر منافذ بین بینی و گلو باریک یا مسدود شده باشد یا غضروف نای بد شکل باشد، این امر میتواند در تنفس و بلع اختلال ایجاد کند. افراد مبتلا به این انسدادها ممکن است دچار عفونتهای دستگاه تنفسی فوقانی، آپنه خواب (sleep apnea) و سوء تغذیه شوند.
سندرم آپرت چندین ناهنجاری مشخصه دست و پا دارد. افراد مبتلا میتوانند انگشتان کوتاه و شستهای پهن و انگشتان بزرگی داشته باشند که به سمت بیرون منحرف میشوند. همچنین ممکن است فیوژن جزئی تا کامل (سینداکتیلی یا syndactyly) انگشتان دست و پا خاص داشته باشند. بسیاری از افراد مبتلا به هم آمیختگی کامل استخوانهای انگشت دوم تا چهارم و یک ناخن منفرد و پیوسته (سینداکتیلی یا syndactyly) دارند.
با این حال، همجوشیهای دیگر نیز ممکن است رخ دهد. مفاصل انگشتان در حدود چهار سالگی سفت میشوند. در پاها، سینداکتیلی معمولاً انگشتان دوم، سوم و چهارم را نیز درگیر میکند. ناخنهای پا ممکن است تا حدی پیوسته یا مجزا باشند. به طور کلی، اندام فوقانی شدیدتر از اندام تحتانی تحت تأثیر سندرم آپرت قرار میگیرند.
سندرم آپرت میتواند سایر سیستمهای اندام را نیز تحت تاثیر قرار دهد:
-
اسکلتی
- کاهش سرعت رشد که منجر به کوتاهی قد میشود، علیرغم وزن طبیعی هنگام تولد و طول تولد
- جوش خوردن مهرههای گردن
- همجوشی دو استخوان بازو
- جوش خوردن استخوانهای مچ دست
-
عصبی
- درجات مختلف تاخیر رشد
- ناتوانی ذهنی خفیف تا متوسط: به نظر میرسد ضریب هوشی به عواملی از جمله سن جراحی رفع فشار جمجمه و وجود ناهنجاریهای اضافی مغز بستگی دارد.
- عدم وجود جسم پینه ای (corpus callosum)، بافت فیبری که به نیمکرههای مغزی میپیوندد.
- عدم تشکیل غشاهایی که معمولاً حفرههای مغز را جدا میکنند.
- بزرگ شدن حفره مغز
- ناهنجاریهای بخشهایی از مغز که با سیستم عصبی خودمختار (autonomous nervous system یا ANS) سروکار دارند. ANS عملکردهای خودکار بدن مانند تنفس یا ضربان قلب را کنترل میکند.
- گوشها
- کم شنوایی
- عفونتهای مزمن گوش
-
قلب (cardiac)
- سوراخ(هایی) در دیواره بطن
- آئورت غلبه کننده (Overriding aorta) زمانی ایجاد میشود که آئورت مستقیماً روی یک سوراخ در دیواره بطن قرار گیرد، به جای اینکه روی بطن چپ باشد. در نتیجه، آئورت میتواند حاوی مقداری خون از بطن راست باشد. این امر میزان اکسیژن انتقالی را کاهش میدهد.
-
شکم
- دهانه باریک تر بین قسمت تحتانی معده و قسمت بالایی روده کوچک
- انسداد مری
-
کلیهها و دستگاه ادراری تناسلی
- مقعد خارج از موقعیت
- انسداد واژن
- عدم افتادن بیضهها
- بزرگ شدن کلیهها به دلیل انسداد
علل
سندرم آپرت در اثر تغییر (جهش) در ژن گیرنده فاکتور رشد فیبروبلاست 2 (fibroblast growth factor receptor-2 یا FGFR2) ایجاد میشود. این ژن نقش مهمی در رشد اسکلتی دارد.
ژنها دستورالعملهایی را برای ایجاد پروتئینهایی ارائه میدهند که نقشهای مشخصی در بدن ما دارند. هنگامی که یک جهش در یک ژن رخ میدهد، محصول پروتئینی ممکن است آن طور که باید کار نکند. در سندرم آپرت ، جهش در FGFR2 منجر به عدم ارتباط صحیح این گیرندهها با فاکتورهای رشد فیبروبلاست میشود.
این امر بر تشکیل بخیههای طبیعی در مغز تأثیر میگذارد و میتواند مانع رشد بسیاری از ساختارهای دیگر در بدن شود. این شکل گیری نامناسب همان چیزی است که باعث ناهنجاریهای دیده شده در سندرم آپرت میشود.
تقریباً در تمام بیماران گزارش شده، این اختلال توسط یکی از دو جهش خاص ژن FGFR2 ایجاد شده است. (این جهشها “Ser252Trp” و “Pro253Arg” نامیده میشوند.) این جهشها ممکن است تظاهرات کمی متفاوت از جمله شدت سنداکتیلی ایجاد کنند. جهشهای مختلف در ژن FGFR2 ممکن است باعث چندین اختلال مرتبط دیگر از جمله سندرم فایفر (Pfeiffer syndrome)، سندرم کروزون (Crouzon syndrome) و سندرم جکسون ویس (Jackson-Weiss syndrome) شود.
در بیش از 95 درصد از بیماران، سندرم آپرت ناشی از یک جهش جدید در ژن FGFR2 است. به نظر میرسد این جهشهای جدید به صورت تصادفی و به دلایل ناشناخته (به صورت پراکنده یا sporadically) رخ میدهند. گزارش شده است که موارد پراکنده ممکن است با افزایش سن پدر همراه باشد.
به ندرت، سندرم آپرت به صورت اتوزومال غالب به ارث میرسد. اختلالات ژنتیکی غالب زمانی اتفاق میافتد که تنها یک نسخه از یک جهش برای ایجاد یک بیماری خاص ضروری است. خطر انتقال جهش از والدین مبتلا به فرزند برای هر بارداری 50 درصد است. این خطر برای مردان و زنان یکسان است.
جمعیتهای آسیب دیده
تخمین زده میشود که سندرم آپرت در حدود یک مورد از هر 65000 تولد رخ میدهد. به نظر میرسد مردان و زنان به تعداد نسبتاً مساوی به سندرم آپرت مبتلا هستند. بیش از 300 مورد از زمان توصیف اولیه این اختلال در سالهای 1894 و 1906 گزارش شده است. گزارش شده است که افراد آسیایی بالاترین میزان ابتلا به سندرم آپرت را دارند.
اختلالات با علائم مشابه
علائم اختلالات زیر ممکن است مشابه علائم سندرم آپرت باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد.
سندرم کارپنتر (Carpenter syndrome) یک اختلال ژنتیکی نادر است که با کرانیوسینوستوز، بافته شدن یا همجوشی (سینداکتیلی یا syndactyly) انگشتان دست یا پا و یا انگشتان دست یا انگشتان پای اضافی (پلی داکتیلی یا polydactyly) همراه است. قسمت بالای سر ممکن است به طور غیر معمول مخروطی (آکروسفالی یا acrocephaly) بوده یا سر کوتاه و پهن به نظر برسد (براکی سفالی یا brachycephaly).
علاوه بر این، بخیههای جمجمه اغلب به طور ناهموار به هم متصل میشوند و باعث میشوند سر و صورت از یک طرف به سمت دیگر نامتقارن به نظر برسند. در برخی موارد، ناهنجاریهای فیزیکی اضافی مانند کوتاهی قد، نقص مادر زادی قلب، چاقی خفیف تا متوسط، فتق ناف یا نارسایی بیضهها در نزول به کیسه بیضه در مردان مبتلا وجود دارد.
بسیاری از افراد مبتلا به این اختلال تحت تأثیر ناتوانی ذهنی خفیف تا متوسط قرار دارند. سندرم کارپنتر میتواند در اثر تغییرات در ژن RAB23 یا ژن MEGF8 ایجاد شود. هر دو نوع سندرم کارپنتر به روش اتوزومال مغلوب به ارث میرسند.
سندرم کروزون (Crouzon syndrome) یک اختلال ژنتیکی نادر است که با کرانیوسینوستوزیس همراه است. افراد مبتلا به سندرم کروزون همچنین دارای ناهنجاریهای میانی صورت، چشمهای بیرون زده و انسداد راههای هوایی هستند که منجر به مشکلات تنفسی و بلع میشود. برخی از افراد مبتلا سر بسیار بزرگی دارند (هیدروسفالی). سندرم کروزون معمولاً شامل ناتوانی ذهنی یا مشکلات بازوها، پاها، دستها یا پاها نمیشود. سندرم کروزون در اثر تغییر در یکی از ژنهای FGFR، معمولاً FGFR2، ایجاد میشود و به صورت اتوزومال غالب به ارث میرسد.
سندرم جکسون ویس (JWS یا Jackson-Weiss syndrome) یک اختلال ژنتیکی نادر است که شامل کرانیوسینوستوز و ناهنجاریهای پا است. دامنه و شدت علائم و یافتهها حتی در میان اعضای آسیب دیده یک خانواده بسیار متفاوت است. یافتههای اولیه ممکن است شامل نواحی غیر عادی صاف و توسعه نیافته میانی صورت (هیپوپلازی میانی صورت یا midfacial hypoplasia)، انگشتان بزرگ غیر طبیعی و یا ناهنجاری یا جوش خوردن برخی از استخوانهای داخل پا باشد.
JWS میتواند به صورت پراکنده رخ دهد یا میتواند در الگوی اتوزومال غالب به ارث برسد. انواع خاصی از ژن FGFR2 میتواند باعث ایجاد JWS شود، اگرچه جهش در ژنهای دیگر (مانند FGFR3) میتواند شرایط ظاهری مشابهی ایجاد کند.
سندرم فایفر (Pfeiffer syndrome) یک اختلال ژنتیکی نادر است که با کرانیوسینوستوزیس و انحراف غیر طبیعی شستها و انگشتان بزرگ پا مشخص میشود. اکثر افراد مبتلا همچنین در قسمت میانی صورت (چشمهای بیرون زده) و کم شنوایی هدایت کننده تفاوتهایی دارند. سه شکل از سندرم فایفر وجود دارد. نوع II و III جدی تر هستند. سندرم فایفر یک بیماری اتوزومال غالب همراه با جهش در ژنهای FGFR2 و FGFR1 است.
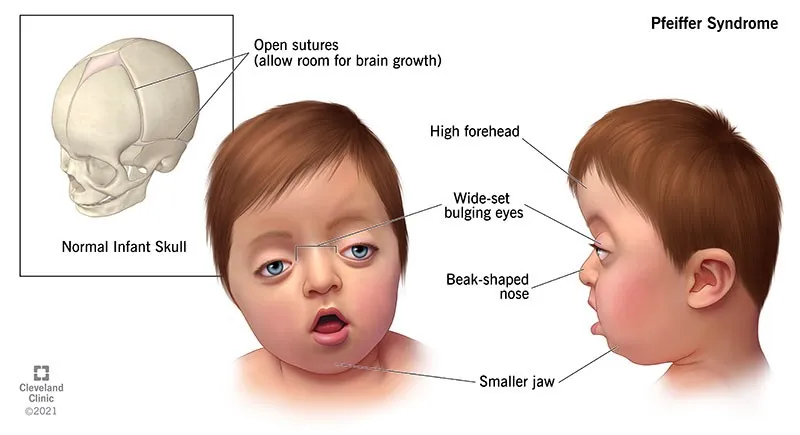
سندرم Saethre-Chotzen (SCS) یکی دیگر از اختلالات ژنتیکی نادر است که با کرانیوسینوستوز و یا تار شدن یا فیوژن (سینداکتیلی) انگشتان دست یا پا مشخص میشود. در بسیاری از بیماران، بخیههای جمجمه ممکن است به طور ناموزون جوش بخورند و باعث شوند که دو طرف سر و صورت نامتقارن به نظر برسند.
تغییرات اضافی در ناحیه جمجمه و صورت نیز ممکن است وجود داشته باشد، مانند چشمهای با فاصله زیاد (هیپرتلوریسم چشمی یا ocular hypertelorism) با حفرههای چشمی کم عمق غیر معمول، افتادگی پلک فوقانی و حالتی که چشمها در یک جهت قرار نمیگیرند (استرابیسم یا strabismus). برخی از افراد مبتلا ممکن است بینی “منقاری یا beaked “، انحراف پارتیشنی که سوراخهای بینی را جدا میکند، گوشهای کوچک و کم شنوا و فک فوقانی توسعه نیافته نیز داشته باشند.
این اختلال همچنین با تغییرات دست و پا، مانند جوش خوردن نسبی بافتهای نرم (سینداکتیلی جلدی یا cutaneous syndactyly) انگشتان دست و پا، دیجیتهای غیر عادی کوتاه و انگشتان بزرگ پهن همراه است. هوش معمولاً طبیعی است. SCS به روش اتوزومال غالب به ارث میرسد.
تشخیص
تشخیص سندرم آپرت اغلب در بدو تولد یا در دوران نوزادی انجام میشود. یک فرد از طریق ارزیابی بالینی و انواع آزمایشات تخصصی تشخیص داده میشود. ویژگیهای فیزیکی مانند ناهنجاریهای صورت یا سینداکتیلی شناسایی میشوند.
ناهنجاریهای اسکلتی و نقایص مادرزادی قلب ممکن است با استفاده از تصویر برداری، مانند سی تی اسکن یا MRI شناسایی شوند. اختلال شنوایی ممکن است در طی آزمایش غربالگری شنوایی نوزاد تشخیص داده شود. افراد همچنین ممکن است آزمایشی برای جهش در ژن FGFR2 انجام دهند که میتواند تشخیص ژنتیکی سندرم آپرت را ارائه دهد.
در برخی موارد، ویژگیهای سندرم آپرت ممکن است قبل از تولد تشخیص داده شود. این کار از طریق سونوگرافی دو بعدی یا سه بعدی قبل از تولد یا تصو یربرداری رزونانس مغناطیسی (magnetic resonance imaging یا MRI) انجام میشود.
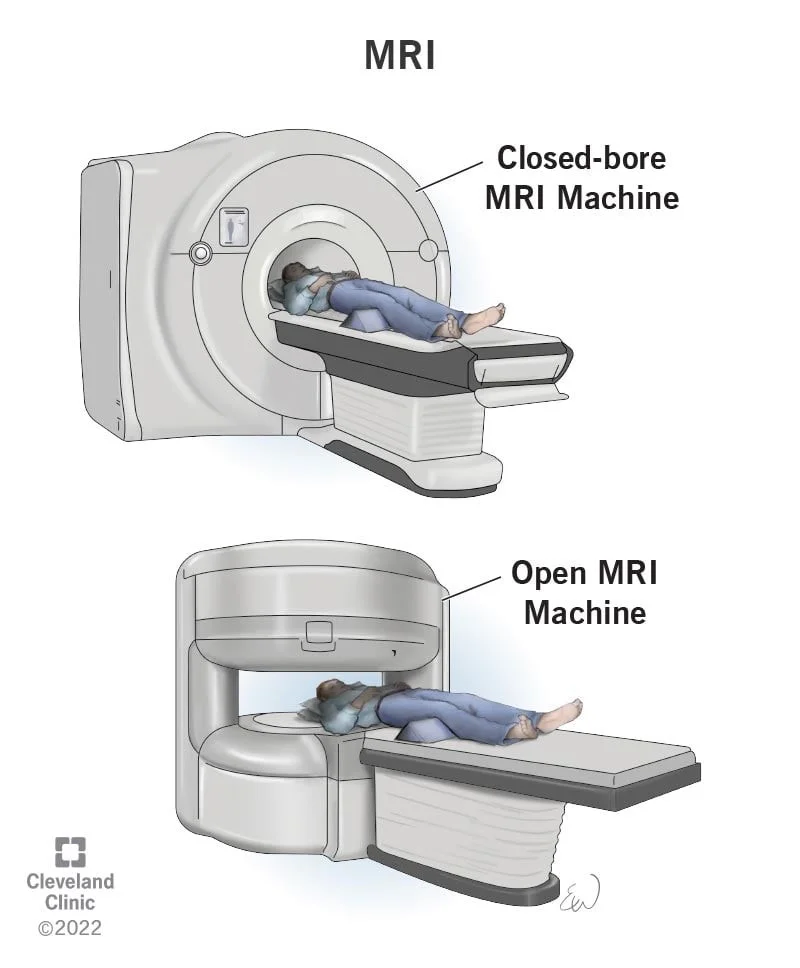
سونوگرافی یک روش غیر تهاجمی است که میتواند تصویری از جنین را ببیند. این میتواند تفاوت در شکل جمجمه، ناهنجاریهای صورت و سینداکتیلی را تشخیص دهد. ام آر آی جنین میتواند جزئیات بیشتری از مغز جنین را نسبت به سونوگرافی ارائه دهد.
درمان
درمان سندرم آپرت بر اساس اینکه چه علائمی در فرد دیده میشود، متفاوت است. چنین درمانی ممکن است به مراقبت توسط تیمی از ارائه دهندگان مراقبتهای بهداشتی از جمله پزشکان اطفال و جراحان نیاز داشته باشد. متخصصان ممکن است شامل متخصصان شنوایی، جراحان مغز و اعصاب، پزشکان متخصص در اختلالات اسکلت، مفاصل و ماهیچهها (ارتوپد)، پزشکانی که در اختلالات گوش، بینی و گلو تخصص دارند (متخصص گوش و حلق و بینی) و پزشکانی که در ناهنجاریهای قلبی تخصص دارند (متخصصان قلب) باشند.
درمانهای خاص برای سندرم آپرت علامتی و حمایتی هستند. کرانیوسینوستوز و هیدروسفالی ممکن است منجر به افزایش غیر طبیعی فشار داخل جمجمه و مغز شود. در چنین مواردی، جراحی زود هنگام (در عرض 2 تا 4 ماه پس از تولد) ممکن است برای اصلاح کرانیوسینوستوز توصیه شود. برای مبتلایان به هیدروسفالی، جراحی ممکن است شامل قرار دادن یک لوله (شانت یا shunt) برای تخلیه مایع مغزی نخاعی اضافی (CSF یا cerebrospinal fluid) از مغز باشد. CSF به قسمت دیگری از بدن تخلیه میشود که در آنجا قابل جذب باشد.
جراحی اصلاحی و ترمیمی ممکن است برای کمک به اصلاح ناهنجاریهای جمجمه صورت توصیه شود. جراحی همچنین ممکن است بتواند به اصلاح پلی داکتیلی و سینداکتیلی و سایر نقایص اسکلتی یا ناهنجاریهای فیزیکی کمک کند. برای افرادی که دارای نقص مادرزادی قلب هستند، ممکن است درمان با برخی داروها، مداخله جراحی و یا اقدامات دیگر ضروری باشد. برای برخی از افراد مبتلا به آسیب شنوایی، سمعک ممکن است مفید باشد.
مداخله زود هنگام ممکن است برای اطمینان از اینکه کودکان مبتلا به سندرم آپرت به پتانسیل کامل خود میرسند، مهم باشد. خدمات ویژه مانند فیزیوتراپی، کار درمانی و آموزش ویژه ممکن است مفید باشد.
مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود. یک مشاور ژنتیک میتواند علل سندرم آپرت را توضیح دهد. آنها همچنین میتوانند در مورد شانس داشتن فرزندان بیشتر مبتلا به سندرم آپرت صحبت کنند. حمایت روانی اجتماعی برای کل خانواده نیز ضروری است.
همچنین بخوانید:
- سندرم کروزون چیست؟
- مایع مغزی نخاعی یا CSF چیست؟
- سندرم X شکننده (Fragile X Syndrome)
- سندرم Singleton-Merten چیست؟ علائم، علل، تشخیص و درمان
مترجم: فاطمه فریادرس
فرق بین سندرم آپرت وفایفر برای تشخیص
هر دو سندرم از گروه کرانیوسینوستوزها هستند ولی تفاوتهای تشخیصی مهم دارند:
سندرم آپرت (Apert):
چسبندگی شدید انگشتان دست و پا (سینداکتیلی)
درگیری صورت واضحتر
معمولاً جهش FGFR2
سندرم فایفر (Pfeiffer):
انگشتان پهن و شستهای بزرگ
سینداکتیلی شدید ندارد
جهش در FGFR1 یا FGFR2
بنابراین مهمترین معیار تشخیص بالینی:
آپرت = انگشتان به هم چسبیده
فایفر = انگشتان پهن و جدا.