



دیسپلازی آکرومزوملیک چیست؟
دیسپلازی آکرومزوملیک یک اختلال اسکلتی بسیار نادر، ارثی و پیشرونده است که منجر به شکل خاصی از کوتاهی قد میشود که به نام کوتولگی اندام کوتاه (short-limb dwarfism) شناخته میشود. این اختلال با آکروملیا (acromelia) و مزوملیا (mesomelia) مشخص میشود. مزوملیا کوتاه شدن استخوانهای ساعد و ساق پا را نسبت به قسمتهای بالایی آن اندامها توصیف میکند.
آکروملیا کوتاه شدن استخوانهای دست و پا است. بنابراین کوتاهی قد افراد مبتلا نتیجه کوتاهی غیرعادی ساعد و کوتاه شدن غیرطبیعی استخوانهای ساق پا، دستها، انگشتان دست، پاها و انگشتان بسیار کوتاه است. این یافتهها در سالهای اول زندگی آشکار میشوند.
مترادفها
- کوتولگی آکرومزومیک (acromesomelic dwarfism)
زیر بخشها
- دیسپلازی آکرومزولیک، نوع ماروتو (Maroteaux type)
- دیسپلازی آکرومزوملیک، نوع Osebold-Remondini
- دیسپلازی آکرومزوملیک با ناهنجاریهای تناسلی (acromesomelic dysplasia with genital anomalies)
- هیپوپلازی فیبولار (fibular hypoplasia) و براکیداکتیلی کمپلکس یا complex brachydactyly (سندرم دو پان یا Du Pan syndrome)
- دیسپلازی گرب یا Grebe dysplasia (شامل نوعهانتر تامپسون یا Hunter-Thompson type)
علائم و نشانههای دیسپلازی آکرومزوملیک
دیسپلازی آکرومزوملیک (AMD) با مهار رشد برخی از استخوانهای بلند (مانند استخوانهای ساعد و ساق پا) مشخص میشود. در نتیجه، افراد مبتلا ساعد و ساق پا کوتاه و قد کوتاه (short-limbed dwarfism) را نشان میدهند. این یافتهها معمولاً در سالهای اول زندگی آشکار میشوند. رشد غیر طبیعی غضروف و استخوان بر سایر استخوانها، به ویژه استخوانهای دست و پا (مانند متاکارپ یا metacarpals، فالانژ یا phalanges، متاتارس یا metatarsals) نیز تأثیر میگذارد.
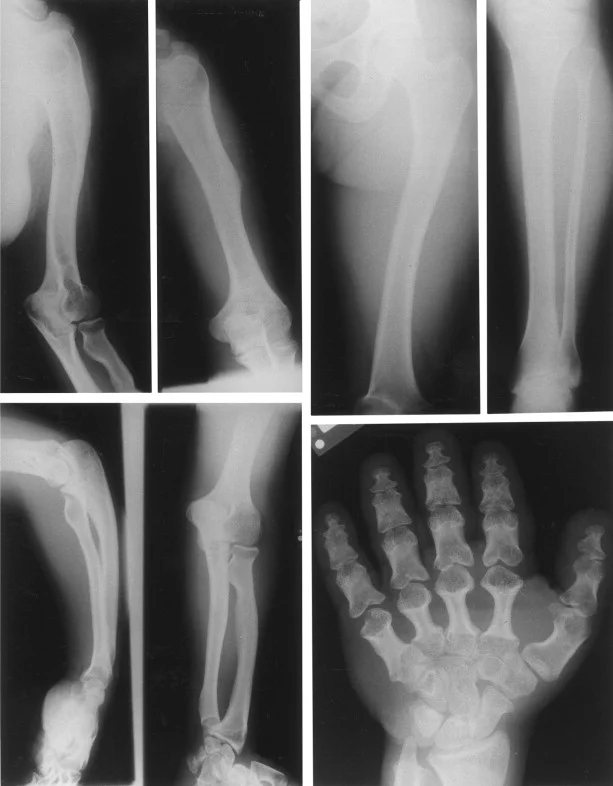
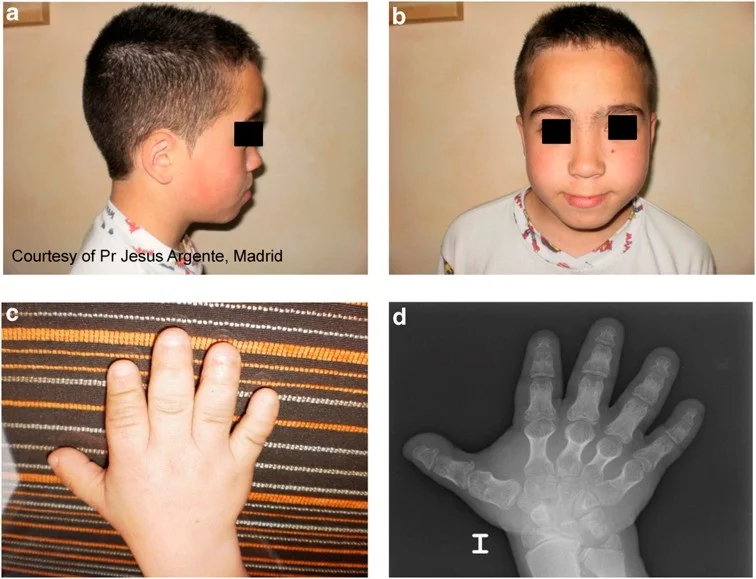
نوزادان مبتلا به دیسپلازی آکروموزومیک اغلب وزن طبیعی هنگام تولد دارند. در بیشتر موارد، علاوه بر داشتن دستها و پاهای کوتاه و غیرمعمول، نوزادان مبتلا اغلب دارای ناهنجاریهای مشخصهای در صورت هستند که در بدو تولد آشکار میشوند. چنین ویژگیهایی ممکن است شامل سر نسبتا بزرگ شده (ماکروسفالی یا macrocephaly)، پیشانی غیرمعمول برجسته (برجستگی جلویی یا frontal bossing) و قسمت پشت برجسته سر (برجستگی پس سری یا occipital prominence) وسط صورت کمی پهن شده و یا یک بینی غیر طبیعی کوچک و پاگ باشد.
در سالهای اول زندگی، از آن جایی که ساعدها، ساق پاها، دستها و پاها متناسب با بقیه بدن رشد نمیکنند، کوتاهی قد (short-limbed dwarfism) شروع به آشکار شدن میکند. به دلیل رشد غیر طبیعی و جوش خوردن زودرس (استخوانی شدن) قسمتهای رشد و شفت استخوانهای بلند بازو، ممکن است استخوانهای قسمت بیرونی و سمت شست ساعد (به ترتیب اولنا یا ulna و رادیوس یا radius) به طور قابل توجهی کوتاه شوند (هیپوپلاستیک) و انحنای غیرعادی داشته باشند.
علاوه بر این، قسمت انتهایی رادیوس (که معمولاً به استخوان بازو میرسد و بخشی از مفصل آرنج را تشکیل میدهد) ممکن است به طور کامل یا جزئی در رفته باشد (subluxation). این وضعیت بدشکلی مادلونگ (Madelung deformity) شناخته میشود.
در نتیجه، افراد مبتلا ممکن است نتوانند به طور کامل بازوهای خود را دراز کنند، بازوها را طوری بچرخانند که کف دستها رو به پایین باشد (پروناسیون یا pronation) یا بازوهای خود را طوری بچرخانند که کف دستها به سمت بالا باشد (سوپینیشن یا supination). برخی از افراد مبتلا ممکن است انحطاط پیشرونده، سفتی، حساسیت و درد آرنج (استئوآرتریت یا osteoarthritis) را نیز تجربه کنند.
دستها و پاها در بدو تولد به طور غیر معمول کوتاه و پهن به نظر میرسند. از آن جایی که ناهنجاریهای غضروف و رشد استخوان در دستها و پاها نیز پیشرونده است، استخوانهای داخل انگشتان دست و پا (فالانژها یا phalanges) و همچنین در بدن دستها (متاکارپالها یا metacarpals) و پاها (متاتارسال یا metatarsals) به طور فزایندهای کوتاهتر میشوند و این حالت در سالهای اول زندگی گسترده تر است.
در طول سال دوم زندگی، انتهای در حال رشد این استخوانها (اپیفیزها یا epiphyses) ممکن است به شکل غیرعادی مانند مخروط یا مربع ظاهر شوند و ممکن است پیش از موعد جوش بخورند. بنابراین، انگشتان دست و پا کوتاه و عجیب به نظر میرسند (براکیداکتیلی یا brachydactyly). دستها و پاها ممکن است به طور غیرمعمول کوتاه، پهن و مربعی به نظر برسند و پاها ممکن است به طور غیر طبیعی صاف به نظر برسند.
در بسیاری از افراد، انگشتان بزرگ پا ممکن است در مقایسه با انگشتان دیگر نسبتا بزرگ به نظر برسند. علاوه بر این، ناخنهای دست و پا نیز ممکن است به طور غیر طبیعی کوتاه و پهن به نظر برسند، اگرچه در غیر این صورت طبیعی هستند. در اوایل کودکی، پوست اضافی و شل (زائد) روی انگشتان ممکن است ایجاد شود.
در اوایل دوران کودکی، افراد مبتلا به AMD ممکن است شروع به نشان دادن ناهنجاریهای استخوانهای ستون فقرات (مهرهها) و انحنای پیشرونده و غیرطبیعی ستون فقرات کنند. کودکان مبتلا ممکن است انحنای غیرعادی از جلو به عقب در قسمت مرکزی ستون فقرات (کیفوز پایین قفسه سینه یا low thoracic kyphosis) و یا انحنای غیرطبیعی اغراق آمیز به سمت پایین ستون فقرات (هیپرلوردوز کمری یا lumbar hyperlordosis) را نشان دهند.
در موارد نادر، ناهنجاریهای اضافی ممکن است وجود داشته باشد. به عنوان مثال، برخی از افراد مبتلا به AMD بلوغ تاخیری را تجربه کردند و در چند مورد گزارش شده، کودکان مبتلا تیرگی قرنیه را نشان دادند.
علل دیسپلازی آکرومزوملیک
تصور میشود که پنج نوع دیسپلازی آکروموزومیک وجود دارد. هر کدام بسیار نادر هستند و هر کدام به عنوان یک صفت ژنتیکی اتوزومال مغلوب به ارث میرسند، به جز نوع AMD Osebold-Remondini که به نظر میرسد اتوزومال غالب است. نوع Maroteaux در کروموزوم 9 در مکان نقشه ژن 9p13-12 ردیابی شده است.
دیسپلازی گرب (شامل نوع AMD هانتر تامپسون) و سندرم دو پان هر کدام به کروموزوم 20 در مکان نقشه ژن 20q11.2 نگاشت شده اند. دیسپلازی آکرومزولیک با ناهنجاریهای تناسلی به 4q23-24 نشان داده میشود. نوع Osebold-Remondini هنوز از نظر ژنتیکی نقشه برداری نشده است.
مطالعات ژنتیکی نشان میدهد که تغییر (جهش) در کروموزوم 9p13-12 (نوع AMD Maroteaux) در ژنی است که پروتئینی را که بر رشد استخوان تأثیر میگذارد، گیرنده پپتید ناتریورتیک B (NPR-B) کد میکند. این یک گیرنده (پروتئینی که به پروتئین دیگری متصل میشود) برای هورمونی به نام پپتید ناتریورتیک (natriuretic peptide) نوع C است، هورمونی که برای رشد استخوان بسیار مهم است.
ژن واقع در کروموزوم 20q11.2 (Grebe dysplasia) پروتئینی را کد میکند که به نام فاکتور رشد و توسعه-5 (GDF5، که قبلاً پروتئین مورفوژنتیک مشتق از غضروف-1، CDMP1 نامیده میشد) شناخته میشود. ژن واقع در کروموزوم 4q23-24 (AMD با ناهنجاریهای تناسلی) پروتئینی به نام گیرنده پروتئین مورفوژنتیک استخوان، نوع B۱ (BMPR1B) را کد میکند. این یک گیرنده برای GDF5 است.
کروموزومهایی که در هسته سلولهای انسانی وجود دارند، حامل اطلاعات ژنتیکی هر فرد هستند. سلولهای بدن انسان به طور معمول دارای 46 کروموزوم هستند. جفت کروموزومهای انسان از 1 تا 22 شماره گذاری شده و کروموزومهای جنسی X و Y تعیین میشوند. مردان دارای یک کروموزوم X و Y و زنان دارای دو کروموزوم X هستند.
هر کروموزوم دارای یک بازوی کوتاه به نام “p” و یک بازوی بلند با نام “q” است. کروموزومها به نوارهای متعدد بیشتری تقسیم میشوند که شماره گذاری میگردند. به عنوان مثال، “کروموزوم 9p13-12” به ناحیه ای در بازوی کوتاه کروموزوم 9 بین باندهای 13 و 12 اشاره دارد. نوارهای شماره گذاری شده محل هزاران ژن موجود در هر کروموزوم را مشخص میکنند.
بیماریهای ژنتیکی با ترکیب ژنهای یک صفت خاص که روی کروموزومهای دریافتی از پدر و مادر قرار دارند، تعیین میشوند.
اختلالات ژنتیکی مغلوب زمانی اتفاق میافتد که یک فرد دو نسخه از یک ژن غیر طبیعی را برای یک صفت به ارث میبرد، یکی از هر یک از والدین. اگر فردی یک ژن طبیعی و یک ژن برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود اما معمولا علائمی از خود نشان نمیدهد. خطر انتقال ژن معیوب و داشتن فرزند مبتلا برای دو والدین ناقل در هر بارداری 25 درصد است.
خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس اینکه کودک از هر دو والدین ژن طبیعی دریافت کند و از نظر ژنتیکی برای آن صفت خاص طبیعی باشد 25 درصد است. این خطر برای مردان و زنان یکسان است.
همه افراد حامل 4-5 ژن غیر طبیعی هستند. والدینی که از خویشاوندان نزدیک (فامیلی) هستند، شانس بیشتری نسبت به والدین غیر خویشاوند دارند که هر دو دارای ژن غیرطبیعی مشابهی باشند که خطر بچه دار شدن با اختلال ژنتیکی مغلوب را افزایش میدهد.
اختلالات ژنتیکی غالب زمانی اتفاق میافتد که تنها یک نسخه از یک ژن غیر طبیعی برای ایجاد یک بیماری خاص ضروری باشد. ژن غیر طبیعی میتواند از هر یک از والدین به ارث برسد یا میتواند نتیجه یک جهش جدید (تغییر ژن) در فرد مبتلا باشد. خطر انتقال ژن غیر طبیعی از والدین مبتلا به فرزندان 50 درصد برای هر بارداری است. این خطر برای مردان و زنان یکسان است.
جمعیتهای آسیب دیده
از سال 2005، حدود 10 فرد مبتلا به ADM نوع Hunter-Thompson و حدود 40 تا 50 بیمار از نوع Maroteaux AMD در متون پزشکی گزارش شده بودند. تعداد موارد ADM نوع Grebe مشخص نیست اما اعتقاد بر این است که این نوع تقریباً به طور کامل محدود به افرادی است که در برزیل زندگی میکنند.
اختلالات با علائم مشابه دیسپلازی آکرومزوملیک
آکندروپلازی شایع ترین اختلال کوتولگی اندام کوتاه است. افراد مبتلا دستها و پاهای بسیار کوتاهی دارند، در حالی که اندازه تنه تقریباً طبیعی است. در طول رشد جنین و دوران کودکی، غضروف به طور معمول به استخوان تبدیل میشود، به جز در چند مکان، مانند بینی و گوش. در افراد مبتلا به آکندروپلازی، در طول این فرآیند مشکلی رخ میدهد، به خصوص در استخوانهای بلند (مانند استخوانهای بالای بازو و ران).
سرعت تبدیل سلولهای غضروفی در صفحات رشد استخوانهای بلند به استخوان آهسته است و منجر به کوتاهی استخوان و کاهش قد میشود. این سندرم به دلیل جهشهای خاص در ژن گیرنده فاکتور رشد فیبروبلاست 3 (fibroblast growth factor receptor 3 یا FGFR3) ایجاد میشود.
تفاوت آکندروپلازی با AMD در این است که استخوانهای فوقانی بازوها و پاها (بازو و استخوان ران) بیشترین آسیب را در آکندروپلازی میبینند، در حالی که استخوانهای تحتانی (رادیوس و اولنا در بازو، درشت نی و نازک نی در ساق پا) و دستها و پاها که بیشترین آسیب را در AMD دارند.
آکرودیسوستوز (Acrodysostosis) یک اختلال بسیار نادر است که با استخوانهای ناهنجار و کوتاه دست و پا (دیزوستوز محیطی یا peripheral dysostosis)، کوتاهی غیرطبیعی انگشتان دست و پا (براکیداکتیلی یا brachydactyly)، ناهنجاری و کوتاه شدن استخوانهای ساعد (رادیوس و اولنا) و تاخیر رشد پیشرونده مشخص میشود. منجر به کوتولگی اندام کوتاه میشود.
با بزرگتر شدن کودکان مبتلا به این اختلال، ممکن است حرکات دستها، پاها و یا آرنجها به تدریج دچار اختلال و محدودیت شده و همچنین درد و تورم در مفاصل مختلف بدن (آرتریت یا arthritis) را تجربه کنند.
افراد مبتلا همچنین ناهنجاریهای مشخصهای در ناحیه سر و صورت (جمجمه صورت) از جمله بینی غیر طبیعی صاف، توسعه نیافته (هیپوپلاستیک یا hypoplastic)، استخوان فک بالا (هیپوپلازی فک بالا یا maxillary hypoplasia)، چشمهای با فاصله زیاد (هیپرتلوریسم چشم یا ocular hypertelorism) و یا چینهای اضافی پوست که ممکن است تا حدی گوشههای داخلی چشم را بپوشاند (چینهای اپیکانتال یا epicanthal folds)، نشان میدهند.
ممکن است ناتوانی ذهنی نیز وجود داشته باشد. در بیشتر موارد تصور میشود که آکرودیسوستوز به دلایل ناشناخته (پراکنده یا sporadic) تصادفی رخ میدهد.
دیسپلازی آکرومیکریک یکی دیگر از اختلالات ارثی بسیار نادر است که با دست و پاهای غیر طبیعی کوتاه، تاخیر در رشد و بلوغ تاخیری استخوان که منجر به کوتولگی اندام کوتاه و ناهنجاریهای خفیف صورت میشود، مشخص میشود.
ناهنجاریهای جمجمه صورتی ممکن است شامل یک سوراخ غیر طبیعی باریک بین پلکهای بالا و پایین (شکافهای کف دست) و یک بینی کوتاه با سوراخهای بینی رو به بالا باشد. در اغلب موارد، به نظر میرسد دیسپلازی آکرومیکریک به صورت تصادفی، به دلایل ناشناخته (به صورت پراکنده) رخ میدهد. با این حال، توارث اتوزومال غالب رد نشده است.
چندین سندرم کوتاه شدن استخوانهای دست و پا وجود دارد که به نام براکیداکتیلی (brachydactyly) شناخته میشوند. دو مورد از این سندرمها، براکیداکتیلی نوع A2 و C نیز در اثر جهش در ژن GDF5 ایجاد میشوند.
کوتاهی قد ممکن است بیان طبیعی پتانسیل ژنتیکی باشد که در این صورت سرعت رشد طبیعی است یا ممکن است در نتیجه شرایطی باشد که باعث شکست رشد با نرخ رشد کمتر از حد طبیعی میشود. شکست رشد (Growth failure) اصطلاحی است که نرخ رشد کمتر از سرعت رشد مناسب برای سن را توصیف میکند.
اگر کودکی قدش کمتر از صدک پنجم (fifth percentile) باشد، کوتاه قد است. در عوض، برخی کوتاهی قد را به عنوان قد کمتر از 2 انحراف استاندارد زیر میانگین تعریف میکنند که نزدیک به صدک سوم است. بنابراین، 3-5 درصد از همه کودکان کوتاه قد در نظر گرفته میشوند. بسیاری از این کودکان در واقع سرعت رشد طبیعی دارند.
این کودکان کوتاه قد شامل کودکانی هستند که قد کوتاه خانوادگی یا تاخیری در رشد و بلوغ دارند. برای حفظ صدک قد یکسان در نمودار رشد، سرعت رشد باید حداقل در صدک 25 باشد. هنگامی که همه کودکان با قد کوتاه را در نظر میگیریم، فقط تعداد کمی از آنها واقعاً دارای یک تشخیص خاص قابل درمان هستند، مانند کمبود هورمون رشد یا کم کاری تیروئید. اکثر این کودکان دارای سرعت رشد آهسته هستند.
تشخیص دیسپلازی آکرومزوملیک
در اکثر بیماران، دیسپلازی آکرومزوملیک در چند سال اول زندگی بر اساس ارزیابی بالینی کامل، شرح حال دقیق بیمار، شناسایی یافتههای مشخصه و تکنیکهای تصویر برداری پیشرفته تشخیص داده میشود.
اگرچه ممکن است دستها و پاها در بدو تولد بهطور غیرمعمول کوتاه و پهن به نظر برسند اما ناهنجاریهای پیشرونده مرتبط با این اختلال (مانند کوتاه شدن غیر طبیعی استخوانهای ساعد و ساق پا و کوتاهی قد، کوتاه شدن و پهن شدن بیشتر استخوانهای دست و پا، ناهنجاریهای پیشرونده مهرهای، محدود شدن بازو آرنج و بازو و غیره) معمولاً تا اواخر دوران نوزادی یا اوایل کودکی آشکار نمیشوند.
مطالعات تخصصی اشعه ایکس ممکن است رشد غیر طبیعی و همجوشی زودرس نواحی را تأیید کند که در آن شفت (دیافیز) برخی از استخوانهای بلند (یعنی استخوانهای بازوها و پاها) به انتهای رشد آنها (اپیفیز) میرسند. علاوه بر این، آنها ممکن است همجوشی غیرطبیعی انتهای در حال رشد استخوانها را در انگشتان، انگشتان پا، دستها و پاها (مانند فالانژها، متاکارپالها، متاتارسها) نشان دهند.
چنین مطالعاتی همچنین ممکن است وجود و یا میزان ناهنجاریهای استخوانی ناشی از آن را تأیید کند (مانند استخوان زند کوتاه و رادیوس، سر رادیال دررفته یا سابلوکس، فالانژهای کوتاه و بدشکل و غیره) و همچنین سایر ناهنجاریهای اسکلتی که ممکن است با آکرومزولیک همراه باشد. دیسپلازی (مانند ناهنجاریهای مهره ای و در نتیجه کیفوز پایین قفسه سینه و یا هیپرلوردوز کمری، ایلیا هیپوپلاستیک و غیره).
درمان دیسپلازی آکرومزوملیک
درمان دیسپلازی آکرومزوملیک به سمت علائم و ویژگیهای فیزیکی خاص است که در هر فرد آشکار است. درمان ممکن است به تلاش هماهنگ تیمی از متخصصان نیاز داشته باشد. متخصصان اطفال، متخصصانی که ناهنجاریهای اسکلتی را ارزیابی و درمان میکنند (ارتوپد)، فیزیوتراپها و یا سایر متخصصان مراقبتهای بهداشتی ممکن است نیاز داشته باشند که به طور سیستماتیک و جامع برای درمان کودک مبتلا برنامهریزی کنند.
درمانهای اختصاصی برای درمان دیسپلازی آکرومزوملیک علامتی (symptomatic) و حمایتی (supportive) هستند.
انحنای غیر طبیعی ستون فقرات (مانند کیفوز قفسه سینه پایین و یا هیپرلوردوز کمری) ممکن است با ترکیبی از تمرینات و فیزیوتراپی، سایر تکنیکهای حمایتی، بریسها (braces)، گچها و یا در موارد شدید، جراحی اصلاحی درمان شود. فیزیوتراپی، سایر تکنیکهای حمایتی، و یا جراحی ارتوپدی ممکن است به اصلاح برخی از یافتههای خاص مرتبط با دیسپلازی آکرومزولیک کمک کند.
مداخله زود هنگام برای اطمینان از اینکه کودکان مبتلا به دیسپلازی آکرومزوملیک به پتانسیل خود میرسند، مهم است. خدمات ویژه ای که ممکن است برای کودکان آسیب دیده مفید باشد ممکن است شامل حمایت اجتماعی و سایر خدمات پزشکی، اجتماعی و یا حرفه ای باشد.
مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود. درمان دیگر این اختلال علامتی و حمایتی است.
آزمایشات و مطالعات بالینی
هورمون رشد انسانی نوترکیب (rhGH یا Recombinant human growth hormone) در تعداد بسیار کمی از بیماران مبتلا به AMD Maroteaux نوع استفاده شده است و هیچ یک بهبودی در سرعت قد نشان ندادند. از این رو، rhGH برای درمان این اختلالات توصیه نمیشود.
همچنین بخوانید:
مترجم: فاطمه فریادرس