


دستهبندی نشده
تالاسمی بتا: علائم، علل، انواع و درمان
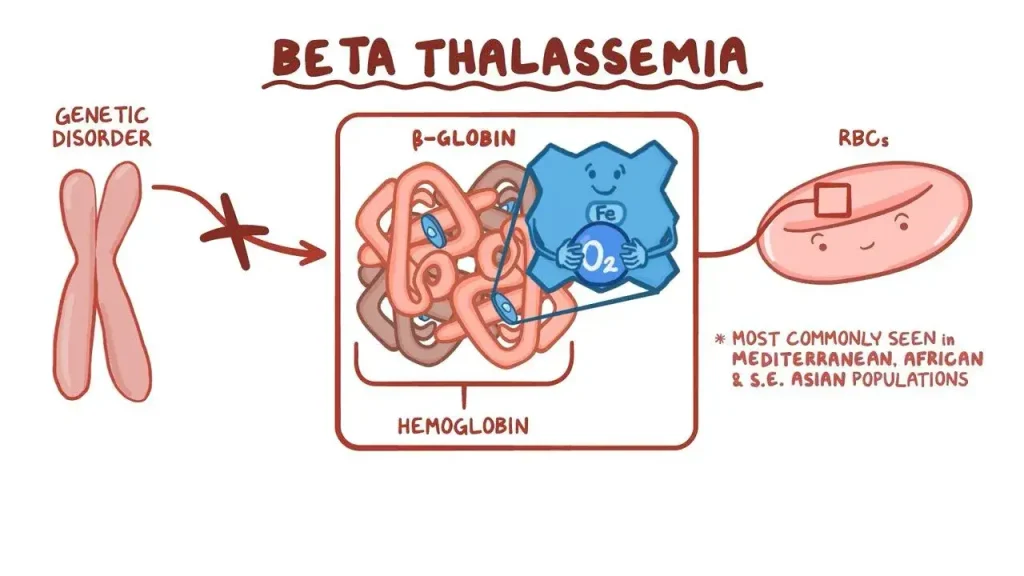
خلاصهای از تالاسمی بتا (Beta Thalassemia)
تالاسمی بتا یک اختلال خونی ارثی است که با کاهش سطح هموگلوبین عملکردی مشخص میشود. هموگلوبین (Hemoglobin) در گلبولهای قرمز خون یافت میشود. این رنگدانه قرمز، غنی از آهن و حامل اکسیژن خون است. وظیفه اصلی گلبولهای قرمز، رساندن اکسیژن به سراسر بدن است.
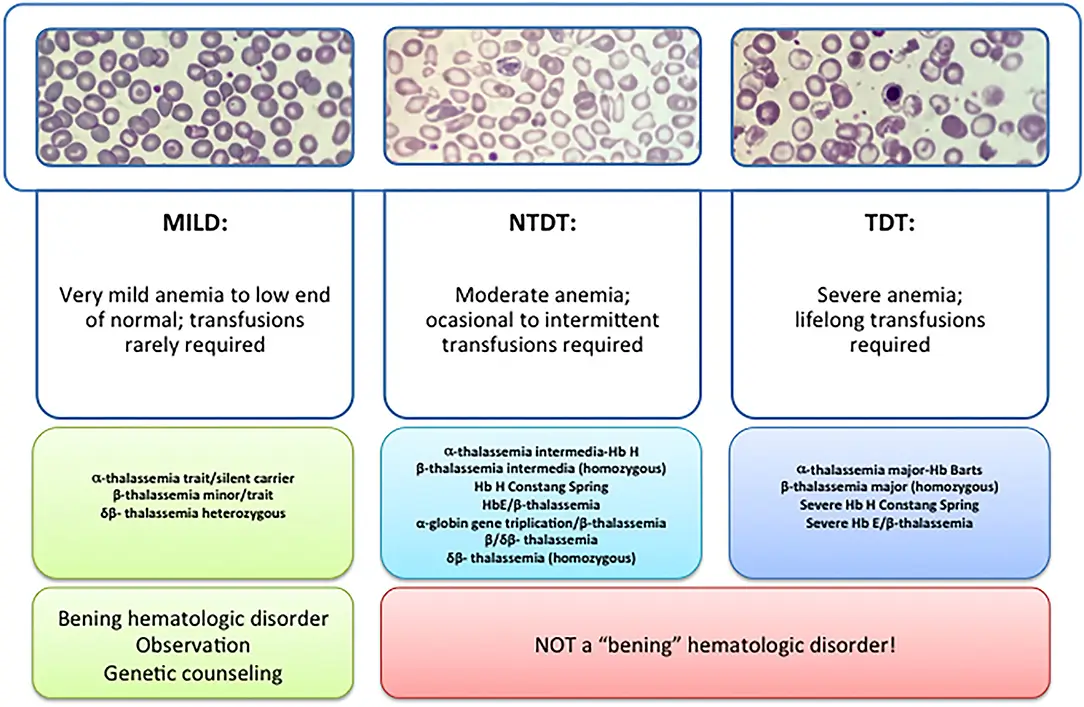
تالاسمی بتا دارای سه شکل اصلی مینور (minor)، میانی (intermedia) و ماژور (major) است که نشان دهنده شدت بیماری است. افراد مبتلا به بتا تالاسمی مینور معمولاً هیچ علامتی ندارند (بدون علامت یا asymptomatic) و افراد اغلب از ابتلای خود به این بیماری اطلاعی ندارند. برخی از افراد کم خونی بسیار خفیف را تجربه میکنند. افراد مبتلا به بتا تالاسمی ماژور بیان شدید این اختلال را دارند. آنها تقریباً همیشه نیاز به تزریق خون منظم و مراقبتهای پزشکی مداوم و مادام العمر دارند.
علائم بتا تالاسمی اینترمدیا به طور گسترده ای متغیر است و شدت آن در محدوده وسیعی بین دو حالت افراطی شکل ماژور و مینور قرار میگیرد. یافته مشخصه تالاسمی بتا کم خونی است که به دلیل کوچک بودن غیر طبیعی گلبولهای قرمز (میکروسیتی یا microcytic)، تولید نشدن در مقادیر طبیعی و نداشتن هموگلوبین عملکردی کافی ایجاد میشود. در نتیجه، افراد مبتلا به اندازه کافی خون غنی از اکسیژن در سراسر بدن دریافت نمیکنند.
افراد مبتلا ممکن است علائم کلاسیک کم خونی از جمله خستگی، ضعف، تنگی نفس، سرگیجه یا سردرد را تجربه کنند. کم خونی شدید در صورت عدم درمان میتواند باعث عوارض جدی و حتی تهدید کننده زندگی شود. افراد مبتلا با تزریق خون منظم درمان میشوند. به دلیل تزریق مکرر خون، افراد مبتلا به تالاسمی بتا ممکن است سطح آهن اضافی در بدن ایجاد کنند (iron overload یا اضافه بار آهن). اضافه بار آهن میتواند علائم مختلفی را ایجاد کند که بر سیستمهای مختلف بدن تأثیر میگذارد اما میتوان آن را با دارو درمان کرد.
تالاسمی بتا در اثر تغییرات (انواع یا جهش) در ژن هموگلوبین بتا (HBB) ایجاد میشود. افراد مبتلا به بتا تالاسمی مینور دارای جهش در یک ژن HBB هستند، در حالی که افراد با فرمهای متوسط و عمده دارای جهش در هر دو ژن HBB هستند.
معرفی
تالاسمی (Thalassemia) اصطلاحی برای گروهی از اختلالات است که در آن سطوح هموگلوبین کاهش یافته، تولید گلبولهای قرمز خون و کم خونی کاهش مییابد. دو شکل اصلی وجود دارد – تالاسمی آلفا (alpha thalassemia) و تالاسمی بتا (beta thalassemia) که هر کدام زیرگروههای مختلفی دارند. بتا تالاسمی مینور که به عنوان صفت بتا تالاسمی (beta thalassemia trait) نیز شناخته میشود، یک بیماری شایع است.
بتا تالاسمی ماژور اولین بار در ادبیات پزشکی در سال 1925 توسط یک پزشک آمریکایی به نام توماس کولی (Thomas Cooley) توصیف شد. بتا تالاسمی ماژور به عنوان کم خونی کولی (Cooley’s anemia) نیز شناخته میشود. امروزه، تصویر بالینی کلاسیک بتا تالاسمی ماژور زمانی که درمان زود هنگام و به طور منظم برای این بیماری آغاز شود، به ندرت دیده میشود.
به دلیل کم خونی و نیاز به تزریق خون، تالاسمی در حال حاضر به عنوان تالاسمی وابسته به انتقال خون (TDT یا transfusion dependent thalassemia) یا تالاسمی غیر وابسته به انتقال خون (non-transfusion dependent thalassemia یا NTDT) به جای مینور، متوسط یا ماژور توصیف میشود.
مترادفها
- کم خونی مدیترانه ای (Mediterranean anemia)
تقسیمات فرعی
- بتا تالاسمی ماژور یا beta thalassemia major (کم خونی کولی یا Cooley’s anemia)
- بتا تالاسمی اینترمدیا (beta thalassemia intermedia)
- بتا تالاسمی مینور یا beta thalassemia minor (ویژگی بتا تالاسمی یا beta thalassemia trait)
- تالاسمی بتا غالب (dominant beta thalassemia)
علائم و نشانهها
علائم و شدت بتا تالاسمی از فردی به فرد دیگر بسیار متفاوت است. افراد مبتلا به بتا تالاسمی مینور علائم این اختلال را ندارند اما ممکن است کم خونی خفیفی داشته باشند. بسیاری از افراد مبتلا به بتا تالاسمی مینور هرگز نمیدانند که حامل ژن تغییر یافته این اختلال هستند.
تشخیص بتا تالاسمی ماژور معمولاً در دو سال اول زندگی انجام میشود و افراد برای زنده ماندن نیاز به تزریق خون منظم و مراقبتهای پزشکی مادام العمر دارند. هنگامی که این اختلال در مراحل بعدی زندگی ایجاد میشود، تشخیص بتا تالاسمی اینترمدیا داده میشود. افراد ممکن است فقط در موارد نادر و خاص نیاز به تزریق خون داشته باشند.
بتا تالاسمی ماژور (beta thalassemia major)
بتا تالاسمی ماژور که با نام کم خونی کولی (Cooley’s anemia) نیز شناخته میشود، شدید ترین شکل بتا تالاسمی است. نوزادان مبتلا علائم را در دو سال اول زندگی، اغلب بین 3 تا 6 ماه پس از تولد، نشان میدهند. “توصیف” کامل یا کلاسیک بتا تالاسمی ماژور عمدتاً در کشورهای در حال توسعه رخ میدهد. بیشتر افراد علائم شدیدی را که در زیر به آنها اشاره میشود، بروز نمیدهند. اگرچه بتا تالاسمی ماژور یک بیماری مزمن و مادام العمر است، اگر افراد از درمانهای توصیه شده فعلی پیروی کنند، بیشتر افراد میتوانند زندگی شاد و رضایت بخشی داشته باشند.
در این بیماری کم خونی شدید ایجاد میشود و با خستگی، ضعف، تنگی نفس، سرگیجه، سردرد و زردی پوست، غشاهای مخاطی و سفیدی چشم (یرقان یا jaundice) همراه است. نوزادان مبتلا معمولاً بر اساس سن و جنسیت (ناتوانی در رشد) رشد کرده و وزن اضافه نمیکنند. برخی از نوزادان به تدریج رنگ پریده (pallor) میشوند. مشکلات تغذیه، اسهال، تحریک پذیری یا گیجی، تبهای مکرر، بزرگ شدن غیر طبیعی کبد (هپاتومگالی یا hepatomegaly) و بزرگ شدن غیر طبیعی طحال (سپلنومگالی یا splenomegaly) نیز ممکن است رخ دهد.
اسپلنومگالی ممکن است باعث بزرگ شدن یا تورم شکم شود. اسپلنومگالی ممکن است با طحال بیش فعال (هیپرسپلنیسم یا hypersplenism) همراه باشد، وضعیتی که میتواند به دلیل تجمع سلولهای خونی و تخریب بیش از حد در طحال ایجاد شود. هایپر اسپلنیسم میتواند به کم خونی در افراد مبتلا به تالاسمی بتا کمک کند و باعث کاهش سطح گلبولهای سفید خون، افزایش خطر عفونت و سطوح پایین پلاکتها شود که میتواند منجر به خونریزی طولانی مدت شود.
در صورت عدم درمان، عوارض اضافی ممکن است ایجاد شود. بتا تالاسمی ماژور میتواند باعث انبساط مغز استخوان، ماده اسفنجی داخل استخوانهای خاص شود. مغز استخوان جایی است که بیشتر سلولهای خونی در بدن تولید میشود. مغز استخوان به دلیل تلاش برای جبران کم خونی مزمن منبسط میشود. این انبساط غیر طبیعی باعث میشود که استخوانها نازک تر، پهن تر و شکننده تر شوند. استخوانهای آسیب دیده ممکن است به طور غیر طبیعی رشد کنند (بدشکلی استخوانی یا bone deformities)، به ویژه استخوانهای بلند بازوها و پاها و برخی استخوانهای صورت.
وقتی استخوانهای صورت تحت تأثیر قرار میگیرند، میتواند منجر به ویژگیهای متمایز صورت از جمله پیشانی غیر طبیعی برجسته (برجستگی پیشانی یا frontal bossing)، استخوانهای پر گونه (برجستگی ملاری برجسته یا prominent malar eminence)، پل فرورفته بینی و رشد بیش از حد (هیپرتروفی یا hypertrophy) فک بالا (maxillae) و در نتیجه نمایان شدن دندانهای بالا شود. استخوانهای آسیب دیده، به ویژه استخوانهای بلند دستها و پاها، خطر شکستگی بیشتری دارند. برخی از افراد ممکن است دچار «زانوهای ضربهای» (genu valgum) شوند، وضعیتی که در آن پاها به سمت داخل خم میشوند به طوری که وقتی فرد ایستاده است، زانوها حتی اگر مچ پاها و پاها اینطور نباشند، لمس میشوند.
حتی در صورت درمان، ممکن است عوارض ایجاد شود، به ویژه تجمع آهن در بدن (اضافه بار آهن یا iron overload). اضافه بار آهن ناشی از تزریق خون مورد نیاز برای درمان افراد مبتلا به بتا تالاسمی ماژور است. علاوه بر این، افراد مبتلا جذب آهن بیشتری از دستگاه گوارش را تجربه میکنند که به اضافه بار آهن کمک میکند (اگرچه این در درجه اول در افراد درمان نشده رخ میدهد).
اضافه بار آهن میتواند باعث آسیب بافتی و اختلال در عملکرد اندامهای آسیب دیده مانند قلب، کبد و غدد درون ریز شود. آهن بیش از حد میتواند به قلب آسیب برساند و باعث ریتم غیر طبیعی قلب، التهاب غشایی (پریکارد یا pericardium) که قلب را میپوشاند (پریکاردیت یا pericarditis)، بزرگ شدن قلب و بیماری عضله قلب (کاردیومیوپاتی متسع یا dilated cardiomyopathy) شود.
درگیری قلب میتواند به عوارض تهدید کننده زندگی مانند نارسایی قلبی تبدیل شود. درگیری کبد میتواند باعث ایجاد اسکار و التهاب کبد و فشار بالای ورید اصلی کبد (فشار خون پورتال یا portal hypertension) شود. درگیری غدد درون ریز میتواند باعث نارسایی برخی غدد مانند تیروئید (کم کاری تیروئید) و در موارد نادر دیابت شیرین شود. اضافه بار آهن همچنین میتواند با تاخیر رشد و شکست یا تاخیر در بلوغ جنسی همراه باشد.
علائم اضافی که ممکن است رخ دهد شامل تودههایی است که به دلیل تولید سلولهای خونی خارج از مغز استخوان (خون سازی خارج مدولار یا extramedullary hematopoiesis) ایجاد میشود.
این تودهها عمدتاً در طحال، کبد، غدد لنفاوی، قفسه سینه و ستون فقرات تشکیل میشوند و به طور بالقوه میتوانند باعث فشرده شدن ساختارهای مجاور و علائم مختلفی شوند. افراد مبتلا ممکن است دچار زخمهای پا، افزایش خطر ایجاد لختههای خون در ورید (ترومبوز وریدی یا venous thrombosis) و کاهش معدنی شدن استخوان و در نتیجه شکننده شدن استخوانهایی شوند که مستعد شکستگی (پوکی استخوان یا osteoporosis) هستند.
بتا تالاسمی اینترمدیا (beta thalassemia intermedia)
افرادی که با بتا تالاسمی اینترمدیا تشخیص داده شده اند، بیان بسیار متنوعی از این اختلال دارند. کم خونی نسبتاً شدید شایع است و افراد مبتلا ممکن است نیاز به تزریق خون دوره ای داشته باشند. هر مورد منحصر به فرد است. علائم شایع عبارت اند از رنگ پریدگی، یرقان، زخم پا، سنگ کیسه صفرا (کوله لیتیازیس یا cholelithiasis) و بزرگ شدن غیر طبیعی کبد و طحال. ناهنجاریهای اسکلتی متوسط تا شدید (همان طور که در بتا تالاسمی ماژور توضیح داده شده است) ممکن است رخ دهد.
تالاسمی بتا غالب (dominant beta thalassemia)
تالاسمی بتا غالب شکل بسیار نادری است که در آن افرادی که دارای یک ژن HBB جهش یافته هستند علائم خاصی مرتبط با بتا تالاسمی را نشان میدهند. افراد مبتلا ممکن است دچار کم خونی خفیف تا متوسط، یرقان و بزرگ شدن غیر طبیعی طحال (سپلنومگالی یا splenomegaly) شوند.
علل
بیشتر موارد بتا تالاسمی ناشی از جهش در ژن HBB است. در موارد بسیار نادر، از دست دادن ماده ژنتیکی (حذف) که شامل ژن HBB است باعث این اختلال میشود. ژنها دستورالعملهایی را برای ایجاد پروتئینهایی ارائه میدهند که نقش مهمی در بسیاری از عملکردهای بدن دارند.
هنگامی که یک جهش ژنی رخ میدهد، محصول پروتئین ممکن است معیوب یا ناکارآمد بوده یا أصلا وجود نداشته باشد. بسته به عملکرد پروتئین خاص، این میتواند بر بسیاری از اندامهای بدن تأثیر بگذارد. افراد مبتلا به بتا تالاسمی مینور دارای جهش در یک ژن HBB هستند و ناقل این اختلال هستند. افراد مبتلا به بتا تالاسمی اینترمدیا یا ماژور دارای جهش در هر دو ژن HBB هستند.
هموگلوبین معمولی از پروتئینهای تخصصی به نام گلوبین تشکیل شده است، به ویژه دو زنجیره آلفا و دو پروتئین زنجیره بتا که به یک حلقه هِم مرکزی (central heme ring) متصل هستند.
ژن HBB زنجیرههای پروتئینی گلوبین بتا را ایجاد میکند (کد میکند). جهش در یک ژن HBB منجر به کاهش یا عدم تولید زنجیرههای بتا از آن ژن میشود. صرف نظر از این، نسخه دوم (بی پیرایه یا unaffected) از ژن HBB به طور طبیعی عمل میکند و پروتئین زنجیره بتا کافی برای جلوگیری از علائم تولید میکند، اگرچه گلبولهای قرمز هنوز به طور غیر طبیعی کوچک هستند و کم خونی خفیف هنوز هم میتواند ایجاد شود.
جهش در دو ژن HBB منجر به کاهش قابل توجهی از زنجیرههای بتا (بتا تالاسمی اینترمدیا) یا فقدان تقریباً کامل زنجیرههای بتا (بتا تالاسمی ماژور) میشود. کاهش یا فقدان زنجیرههای پروتئینی گلوبین بتا منجر به عدم تعادل با زنجیرههای پروتئین آلفا گلوبین تولید شده به طور معمول و در نهایت تشکیل معیوب گلبولهای قرمز خون، کمبود هموگلوبین عملکردی و عدم رساندن مقادیر کافی اکسیژن به بدن میشود.
در افراد مبتلا به تالاسمی بتا غالب، ژن HBB جهش یافته یک نوع بسیار ناپایدار هموگلوبین ایجاد میکند (سنتز میکند). افراد مبتلا تشکیل گلبولهای قرمز خون ناکارآمد دارند (اریتروپوز یا erythropoiesis).
محققان بر این باورند که عوامل دیگری بر شدت بتا تالاسمی ماژور و میانی از جمله ژنهای اصلاح کننده تأثیر میگذارند. ژنهای اصلاحکننده (Modifier genes)، بر خلاف ژنی که باعث تالاسمی بتا میشود، بر شدت بالینی اختلال تأثیر میگذارد. تحقیقات بیشتری برای کشف ژنهای اصلاح کننده مختلف مرتبط با بتا تالاسمی و نقش آنها در ایجاد این اختلال ضروری است.
تالاسمی بتا در الگوی اتوزومال مغلوب به ارث میرسد. اختلالات ژنتیکی مغلوب زمانی رخ میدهد که یک فرد یک ژن غیر طبیعی را از هر یک از والدین به ارث میبرد. اگر فردی یک ژن طبیعی و یک ژن غیر طبیعی برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود اما معمولاً علائمی از خود نشان نخواهد داد.
خطر این که دو والدین ناقل هر دو ژن غیر طبیعی را منتقل کنند و بنابراین فرزندی مبتلا داشته باشند با هر بارداری 25 درصد است. خطر داشتن فرزند ناقل مانند والدین در هر بارداری 50 درصد است. شانس دریافت ژنهای طبیعی از هر دو والدین برای کودک 25 درصد است. این خطر برای مردان و زنان یکسان است.
جمعیتهای تحت تاثیر
تالاسمی بتا در ایالات متحده نسبتا نادر است اما یکی از شایع ترین اختلالات اتوزومال مغلوب در جهان است. میزان بروز موارد علامت دار تقریباً 1 در 100000 نفر در جمعیت عمومی تخمین زده میشود. این اختلال به ویژه در مدیترانه، خاورمیانه، آفریقا، آسیای مرکزی، شبه قاره هند و خاور دور شایع است. افراد در سایر نقاط جهان که خانواده آنها از این مناطق هستند، بیشتر در معرض خطر ابتلا به تالاسمی بتا هستند.
اختلالات با علائم مشابه
علائم اختلالات زیر میتواند شبیه به تالاسمی بتا باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد.
شرایط مختلف علائم و نشانههایی مشابه با تالاسمی بتا دارند. چنین شرایطی شامل کم خونی سیدروبلاستیک (sideroblastic anemias) و کم خونی دیسریتروپوئیتیک مادرزادی (congenital dyserythropoietic anemias) است. برخی از اختلالات سطوح هموگلوبین جنینی را افزایش میدهند مانند لوسمی میلومونوسیتی نوجوانان (juvenile myelomonocytic leukemia) و کم خونی آپلاستیک (aplastic anemia).
تالاسمی بتا همچنین میتواند همراه با سایر ویژگیها به عنوان بخشی از یک سندرم بزرگتر مانند ترومبوسیتوپنی وابسته به X همراه با تالاسمی (X-linked thrombocytopenia with thalassemia) یا بتا تالاسمی-تریکوتیودیستروفی (beta thalassemia-trichothiodystrophy) رخ دهد. تالاسمی بتا نیز ممکن است همراه با اختلال دیگری که در آن یک اختلال در ساختار هموگلوبین وجود دارد (هموگلوبینوپاتی یا hemoglobinopathy) رخ دهد.
این شامل هموگلوبین E (HbE/بتا تالاسمی)، هموگلوبین C (HbC/بتا تالاسمی) و هموگلوبین S (HbS/بتا تالاسمی) است. وضعیتی که شبیه کم خونی داسی شکل (sickle cell anemia) است و گاهی اوقات به عنوان بیماری تالاسمی بتا داسی (sickle beta thalassemia disease) شناخته میشود.
تشخیص
تشخیص بتا تالاسمی بر اساس شناسایی علائم مشخصه، ارزیابی بالینی و انواع آزمایشات تخصصی است. با بتا تالاسمی ماژور، علائم اولیه اغلب در دو سال اول زندگی آشکار میشوند و شامل عدم رشد، شکم متورم و علائم کم خونی هستند. بتا تالاسمی اینترمدیا ممکن است در افرادی که علائم مشابه (و در عین حال خفیفتر) دارند اما در سنین بالاتر، مشکوک باشد.
در بسیاری از ایالتهای ایالات متحده، نوزادان از طریق غربالگری نوزادان مبتلا به اختلال هموگلوبین تشخیص داده میشوند. غربالگری نوزادان یک برنامه بهداشت عمومی است که نوزادان تازه متولد شده را از نظر انواع اختلالات قابل درمان اما آنهایی که به آسانی در بدو تولد آشکار نمیشوند، آزمایش میکند. برنامه غربالگری نوزادان هر ایالت (و اختلالات خاص آزمایش شده) متفاوت است.
تست و کار بالینی
افراد مشکوک به تالاسمی بتا تحت آزمایش خون مانند شمارش کامل خون (complete blood count یا CBC) قرار خواهند گرفت. CBC چندین جزء و جنبههای خون از جمله تعداد، غلظت، اندازه، شکل و بلوغ سلولهای خونی را اندازه گیری میکند.
یک آزمایش خون تخصصی به نام الکتروفورز هموگلوبین (hemoglobin electrophoresis)، انواع مختلف هموگلوبین موجود در خون را اندازه گیری میکند.
CBC برای اندازه گیری میزان هموگلوبین و تعداد و اندازه و شکل گلبولهای قرمز خون انجام میشود که نسبت به افراد عادی از نظر تعداد و اندازه کوچکتر هستند. گلبولهای قرمز نیز ممکن است رنگ پریده (هیپوکرومیک یا hypochromic) و اشکال مختلف (پویکیلوسیتوز یا poikilocytosis) داشته باشند.
توزیع هموگلوبین در گلبولهای قرمز خون در افراد مبتلا به تالاسمی بتا ناهموار است و در زیر میکروسکوپ به سلولها ظاهر هدف مشخصی میدهد. نمونه خون را میتوان برای اندازه گیری میزان آهن موجود در خون (فریتین یا ferritin) آزمایش کرد که اغلب در افراد مبتلا به تالاسمی بتا افزایش مییابد.
آزمایش ژنتیک مولکولی میتواند تشخیص بتا تالاسمی را تایید کند. آزمایش ژنتیک مولکولی میتواند جهشهایی را در ژن HBB که عامل ایجاد این اختلال است شناسایی کند اما فقط به عنوان یک سرویس تشخیصی در آزمایشگاههای تخصصی در دسترس است. آزمایش ژنتیک مولکولی برای تشخیص بتا تالاسمی ضروری نیست و عموماً برای شناسایی بستگان در معرض خطر و بدون علامت، برای کمک به تشخیص قبل از تولد و برای پیشبینی پیشرفت یا شدت بیماری در موارد خاص استفاده میشود.
درمان
افراد مبتلا به بتا تالاسمی ماژور و اینترمدیا از ارجاع به مرکز درمان تالاسمی بهره مند خواهند شد. این مراکز تخصصی مراقبتهای جامعی را برای افراد مبتلا به تالاسمی بتا از جمله تهیه برنامههای درمانی خاص، نظارت و پیگیری افراد مبتلا و مراقبتهای پزشکی پیشرفته ارائه میکنند. درمان در چنین مرکزی تضمین میکند که افراد و اعضای خانواده آنها توسط یک تیم مراقبتهای بهداشتی حرفه ای (پزشکان، پرستاران، فیزیوتراپیستها، مددکاران اجتماعی و مشاوران ژنتیک) با تجربه در درمان افراد مبتلا به تالاسمی بتا مراقبت خواهند شد. مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود. حمایت روانی اجتماعی برای کل خانواده نیز ضروری است.
رویهها و مداخلات درمانی خاص ممکن است بسته به عوامل متعددی، مانند نوع خاص تالاسمی بتا، پیشرفت بیماری، وجود یا عدم وجود علائم خاص، شدت بیماری پس از تشخیص، سن و سلامت عمومی فرد و یا عوامل دیگر متفاوت باشد. تصمیمات مربوط به استفاده از یک رژیم دارویی خاص و یا سایر درمانها باید توسط پزشکان و سایر اعضای تیم مراقبتهای بهداشتی با مشورت بیمار بر اساس مشخصات مورد، بحث کامل در مورد مزایا و خطرات بالقوه، از جمله عوارض جانبی احتمالی و اثرات طولانی مدت، ترجیح بیمار و سایر عوامل مناسب وی اتخاذ شود.
افراد مبتلا به بتا تالاسمی مینور معمولاً علائمی ندارند و نیازی به درمان ندارند. مهم است که افراد مبتلا به بتا تالاسمی مینور به درستی تشخیص داده شوند تا از درمانهای غیر ضروری برای شرایط مشابه مانند کم خونی فقر آهن جلوگیری شود. این افراد نباید به طور معمول مکمل آهن مصرف کنند.
افراد مبتلا به بتا تالاسمی ماژور نیاز به تزریق خون منظم دارند. انتقال خون یک روش رایج است که در آن افراد مبتلا خون اهدایی دریافت میکنند تا سطح هموگلوبین سالم و کارآمد را به خون خود بازگردانند. در طی این روش، خون اهدایی از طریق یک لوله پلاستیکی کوچک که در یک رگ خونی (داخل وریدی) قرار میگیرد، به بدن میرسد.
این روش ممکن است بین 1-4 ساعت طول بکشد. افراد مبتلا به بتا تالاسمی ماژور نیاز به تزریق خون منظم دارند، در موارد شدید هر 2 تا 4 هفته یکبار. افراد مبتلا به بتا تالاسمی اینترمدیا گاهی اوقات نیاز به تزریق خون دارند، مانند زمانی که از بیماری یا عفونت رنج میبرند یا زمانی که قصد انجام عمل جراحی را دارند.
در سال 2019، سازمان غذا و داروی ایالات متحده (FDA) Reblozyl (luspatercept-aamt) را برای درمان کم خونی در بیماران بزرگسال مبتلا به تالاسمی بتا که نیاز به تزریق منظم گلبول قرمز دارند، تایید کرد. این دارو نیاز به تزریق خون منظم را کاهش میدهد اما این بیماری را درمان نمیکند.
برخی از افراد ممکن است با برداشتن جراحی طحال (splenectomy) درمان شوند. بزرگ شدن غیر طبیعی طحال (اسپلنومگالی یا splenomegaly) میتواند باعث درد شدید شود و به کم خونی کمک کند. اسپلنومگالی میتواند باعث کاهش سطح سلولهای خونی (پلاکتها یا platelets) شود که به خون اجازه لخته شدن میدهد.
بزرگ شدن طحال در افراد مبتلا به تالاسمی بتا ممکن است به دلیل افزایش تخریب گلبولهای قرمز، تشکیل سلولهای خونی خارج از مغز استخوان (خون سازی خارج مغز استخوان)، تزریق مکرر خون یا بار بیش از حد آهن رخ دهد. اگر سایر اشکال درمان شکست بخورند، برداشتن طحال ممکن است در نظر گرفته شود. اسپلنکتومی منجر به بهبود علائم خاص مرتبط با بتا تالاسمی شده است. با این حال، این روش جراحی دارای خطراتی است که در هر مورد با فواید آن سنجیده میشود.
به دلیل پیشرفتهای صورت گرفته در درمان تالاسمی بتا در چند سال گذشته، برداشتن طحال به ندرت به عنوان یک درمان برای افراد مبتلا ضروری است.
افراد مبتلا به بتا تالاسمی ماژور و اینترمدیا ممکن است دچار اضافه بار آهن شوند که به دو دلیل رخ میدهد. اول اینکه تزریق خون باعث تجمع آهن اضافی در بدن میشود. دوم اینکه بتا تالاسمی میتواند باعث افزایش جذب آهن رژیم غذایی توسط دستگاه گوارش شود. بدن هیچ راه معمولی برای حذف آهن اضافی ندارد.
در افرادی که به طور منظم تزریق خون دریافت میکنند، اضافه بار آهن در درجه اول به دلیل درمان اتفاق میافتد. آهن بیش از حد باعث علائم مختلفی میشود که بر سیستمهای مختلف بدن تأثیر میگذارد. اضافه بار آهن با داروهایی که آهن اضافی را از بدن خارج میکنند مانند فریپروک (دفریپرون یا deferiprone) و اگزید (دفراسیروکس یا deferasirox) درمان میشود.
درمان عوارض اضافی تالاسمی بتا یا اضافه بار آهن علامتی و حمایتی است. توجه ویژه برای تشخیص زود هنگام و درمان سریع بیماری قلبی (cardiac) که بالقوه با اضافه بار آهن همراه است توصیه میشود. بیماری قلبی مهمترین عارضه تهدید کننده زندگی در افراد مبتلا به تالاسمی بتا است.
آزمایشات و مطالعات بالینی
برخی از افراد مبتلا ممکن است کاندیدای پیوند سلولهای بنیادی خونساز باشند که به طور بالقوه میتواند این اختلال را درمان کند. با این حال، به دلیل خطر عوارض و مرگ و میر، این دارو برای افرادی با عوارض جدی که به درمانهای دیگر پاسخ نداده اند، اختصاص دارد. سلولهای بنیادی خون ساز سلولهای خاصی هستند که در مغز استخوان یافت میشوند و انواع مختلفی از سلولهای خونی (مانند گلبولهای قرمز، پلاکتها) را تولید میکنند. افراد مبتلا به تالاسمی بتا با پیوند سلولهای بنیادی آلوژنیک درمان میشوند.
در طول این نوع پیوند، سلولهای مغز استخوان فرد مبتلا با شیمی درمانی یا پرتو درمانی ریشه کن میشوند و با مغز سالم اهدا کننده، معمولاً از یک عضو خانواده نزدیک، جایگزین میشوند.
با این حال، تنها حدود 20 درصد از افراد مبتلا یک عضو خانواده کاملاً سازگار یا اهدا کننده غیر مرتبط خواهند داشت. پیوند سلولهای بنیادی خون ساز پتانسیل اصلاح ناهنجاری زمینه ای را دارد که باعث بتا تالاسمی میشود. در حالت ایده آل، پیوند سلولهای بنیادی خونساز باید قبل از سن 16 سالگی و قبل از شروع هپاتومگالی، فیبروز پورتال یا اضافه بار آهن انجام شود.
ژن درمانی به عنوان گزینه دیگری برای افراد مبتلا به بتا تالاسمی ماژور در حال بررسی است. در ژن درمانی، ژن غیر طبیعی بیمار با یک ژن طبیعی جایگزین میشود تا امکان تولید آنزیم فعال و جلوگیری از پیشرفت و پیشرفت بیماری فراهم شود. با توجه به انتقال دائمی ژن طبیعی که میتواند آنزیم فعال را در همه مکانهای بیماری تولید کند، این درمان از نظر تئوری میتواند منجر به “درمان” شود. با این حال، در حال حاضر، چندین مشکل فنی وجود دارد که باید قبل از اینکه ژن درمانی به عنوان یک رویکرد جایگزین مناسب مورد حمایت قرار گیرد، برطرف شود.
داروهای خاصی مانند مشتقات 5-آزاسیتیدین (5-azacytidine)، دسیتابین (decytabine) و بوتیرات (butyrate) به عنوان درمانهای بالقوه برای افراد مبتلا به تالاسمی بتا مورد مطالعه قرار میگیرند.
این داروها باعث ایجاد (سنتز) هموگلوبین جنینی میشوند که در جنین در حال رشد و نوزادان تازه متولد شده (قبل از اینکه بدن شروع به تولید هموگلوبین بزرگسالان کند) تولید میشود. این داروها با زنجیرههای پروتئین آلفا اضافی متصل میشوند و در نتیجه عدم تعادل بین زنجیرههای پروتئین آلفا و زنجیرههای پروتئین بتا را کاهش میدهند. تحقیقات بیشتری برای تعیین ایمنی و اثر بخشی طولانی مدت این داروها ضروری است.
هیدروکسی اوره (Hydroxyurea) داروی دیگری است که به تحریک تولید هموگلوبین جنین کمک میکند. این دارو به عنوان درمانی برای افراد مبتلا به بتا تالاسمی اینترمدیا برای افزایش سطح هموگلوبین، کاهش اندازه تودههای خارج مدولاری و بهبود زخمهای پا مورد مطالعه قرار گرفته است. مطالعات بیشتر مانند کارآزماییهای بالینی تصادفی سازی شده و کنترل شده برای تعیین نقش هیدروکسی اوره در درمان بتا تالاسمی مفید خواهد بود.
همچنین بخوانید:
مترجم: فاطمه فریادرس