



خلاصه: بیماری اندرسن (Andersen Disease) متعلق به گروهی از اختلالات ژنتیکی نادر متابولیسم گلیکوژن است که به عنوان بیماریهای ذخیره گلیکوژن (glycogen storage diseases) شناخته میشود.
گلیکوژن (Glycogen) یک کربوهیدرات پیچیده است که برای استفاده بدن به عنوان انرژی به قند ساده گلوکز تبدیل میشود. بیماریهای ذخیرهسازی گلیکوژن (Glycogen storage diseases) با کمبود آنزیمهای خاصی که در متابولیسم گلیکوژن دخیل هستند، مشخص میشود که منجر به تجمع اشکال یا مقادیر غیر طبیعی گلیکوژن در قسمتهای مختلف بدن، به ویژه کبد و ماهیچه میشود.
بیماری اندرسن (GSD IV) چیست؟

بیماری اندرسن به عنوان بیماری ذخیره گلیکوژن (GSD یا glycogen storage disease) نوع IV نیز شناخته میشود. بیماری اندرسن به دلیل فعالیت کم آنزیم شاخه ساز گلیکوژن (glycogen-branching enzyme) ایجاد میشود که منجر به تجمع گلیکوژن غیر طبیعی در کبد، ماهیچه و یا بافتهای دیگر میشود. در اکثر افراد مبتلا، علائم و یافتهها در ماههای اول زندگی آشکار میشود. چنین ویژگیهایی معمولاً شامل عدم رشد و افزایش وزن با سرعت مورد انتظار (شکست در رشد) و بزرگ شدن غیر طبیعی کبد و طحال (هپاتواسپلنومگالی یا hepatosplenomegaly) است.
در چنین مواردی، سیر بیماری معمولاً با اسکار پیشرونده کبدی (هپاتیک یا hepatic) (cirrhosis) و نارسایی کبدی مشخص میشود که منجر به عوارض بالقوه تهدید کننده زندگی میشود. با این حال، در موارد نادر، بیماری کبدی پیشرونده ممکن است ایجاد نشود. علاوه بر این، چندین نوع عصبی-عضلانی بیماری اندرسن توصیف شده است که ممکن است در بدو تولد، اواخر کودکی یا بزرگسالی آشکار شود. این بیماری به عنوان یک صفت اتوزومال مغلوب به ارث میرسد.
معرفی
بیماری اندرسن به نام محققی (DH Andersen) که در ابتدا این بیماری را در سال 1956 توصیف کرد، نامگذاری شده است.
مترادفها
- آمیلوپکتینوز (amylopectinosis)
- گلیکوژنوز اندرسن (Andersen glycogenosis)
- کمبود شاخه ساز (brancher deficiency)
- کمبود آنزیم شاخه ساز (branching enzyme deficiency)
- گلیکوژنوز (glycogenosis) نوع IV
- بیماری ذخیره گلیکوژن (glycogen storage disease) IV
علائم و نشانهها
بیماری اندرسن یک اختلال چند سیستمی است که ممکن است کبد، ماهیچههای ارادی (اسکلتی)، قلب، سیستم عصبی و سایر بافتهای بدن را تحت تاثیر قرار دهد. ماهیت و سیر بیماری ممکن است از جنبههای مختلفی متفاوت باشد، از جمله سن شروع، علائم و نشانههای مرتبط، میزان تجمع غیرطبیعی گلیکوژن در بافتهای مختلف، و اندامهای خاص آسیب دیده.
با این حال، رایج ترین و کلاسیک ترین شکل این بیماری معمولاً با اسکار داخلی پیشرونده (فیبروز یا fibrosis) و تخریب بافت کبد (سیروز یا cirrhosis)، باقی ماندن مناطقی از بافت اسکار غیر فعال و به تدریج اختلال در عملکرد کبد مشخص میشود. در چنین مواردی، بیماری معمولاً در دوران نوزادی یا تا حدود 18 ماهگی آشکار میشود.
علائم و نشانههای اولیه معمولاً شامل ناتوانی در رشد و افزایش وزن با سرعت مورد انتظار (شکست در رشد) و بزرگ شدن غیر طبیعی کبد و طحال (هپاتواسپلنومگالی یا hepatosplenomegaly) است.
سیروز معمولاً باعث افزایش فشار خون در سیاهرگها از طحال و روده به کبد (فشار خون پورتال یا portal hypertension)، تجمع غیر طبیعی مایع در شکم (آسیت یا ascites)، بزرگ شدن وریدهای دیواره مری (واریس مری یا esophageal varices) که ممکن است پاره شود و منجر به سرفه یا استفراغ خونی شود و نارسایی کبد میشود. در برخی موارد، علائم اولیه و یافتههای مرتبط با سیروز ممکن است شامل تغییر رنگ زرد متمایل به پوست، غشاهای مخاطی و سفیدی چشم (یرقان یا jaundice)، سردرگمی ذهنی و یا سایر ناهنجاریها باشد.
به ندرت، سیروز کبدی مرتبط با بیماری اندرسن نیز ممکن است منجر به کاهش غیر طبیعی سطح گلوکز خون (هیپوگلیسمی یا hypoglycemia) شود. در اکثر افراد مبتلا به بیماری کلاسیک آندرسن، بیماری پیشرونده کبد ممکن است تا حدود پنج سالگی منجر به پیوند کبد یا عوارض بالقوه تهدید کننده زندگی شود. با این حال، برخی موارد نادر نیز گزارش شده است که در آن افراد مبتلا به بیماری کبدی غیر پیشرونده مبتلا هستند. در برخی از این موارد، افراد مبتلا به خفیف ممکن است علائم ظاهری نداشته باشند (بدون علامت یا asymptomatic).
چندین نوع عصبی عضلانی بیماری اندرسن نیز در ادبیات پزشکی شرح داده شده است. در بیشتر موارد، ممکن است درگیری اولیه یا منفرد عضلانی در اواخر دوران کودکی، با بیماری اسکلتی و یا عضله قلب (میوپاتی یا myopathy و یا کاردیومیوپاتی یا cardiomyopathy) وجود داشته باشد.
تجمع گلیکوژن غیرطبیعی در عضله اسکلتی ممکن است منجر به ضعف و خستگی عضلانی، عدم تحمل ورزش، تحلیل عضلانی (آتروفی یا atrophy) و یا علائم و یافتههای دیگر شود. در افراد مبتلا به کاردیومیوپاتی، ضعیف شدن عضله قلب ممکن است منجر به کشش و بزرگ شدن (اتساع یا dilation) حفرههای پایینی قلب (بطن یا ventricles) شود.
کاردیومیوپاتی متسع ممکن است به تدریج منجر به تضعیف عملکرد پمپاژ قلب شود و باعث اختلال در توانایی گردش خون کافی برای تامین نیازهای بدن به اکسیژن شود (نارسایی قلبی یا heart failure). علائم و یافتههای مرتبط ممکن است شامل خستگی، تحریک پذیری، مشکلات تغذیه، کمبود اشتها، شکست در رشد، تنگی نفس با تلاش و در نهایت در حالت استراحت، تجمع غیر طبیعی مایع در بافتهای بدن (ادم یا edema)، اختلالات ریتم قلب (آریتمی یا arrhythmias) و عوارض بالقوه تهدید کننده زندگی در برخی موارد باشد.
یک نوع عصبی عضلانی نیز گزارش شده است که در بدو تولد مشهود است. این شکل ممکن است با ادم عمومی (هیدروپس یا hydrops)، کاهش شدید تون ماهیچههای اسکلتی (هیپوتونی یا hypotonia)، ضعف و آتروفی عضلانی، خم شدن یا گسترش مفاصل متعدد در وضعیتهای ثابت مختلف (انقباضات یا contractures) و درگیری عصبی که منجر به عوارض تهدید کننده حیات در اوایل زندگی مشخص شود.
علاوه بر این، یک نوع عصبی عضلانی نادر نیز در بزرگسالان توصیف شده است. این شکل از بیماری، به اصطلاح بیماری بدن پلی گلوکوزان بزرگسالان (adult polyglucosan body disease)، ممکن است با اختلال در عملکرد سیستم عصبی مرکزی و محیطی مشخص شود. سیستم عصبی مرکزی (central nervous system یا CNS) به مغز و نخاع اشاره دارد.
اعصاب محیطی از CNS به ماهیچهها، غدد، پوست، اندامهای حسی و اندامهای داخلی گسترش مییابد. اعصاب محیطی شامل اعصاب حرکتی، اعصاب حسی و اعصاب سیستم عصبی خودمختار است که در عملکردهای غیر ارادی از جمله تنظیم فشار خون، دما و ضربان قلب نقش دارند. در افراد مبتلا به بیماری بدن پلی گلوکوزان بالغ (adult polyglucosan body disease)، علائم و یافتههای مرتبط ممکن است شامل از دست دادن حس در پاها، ضعف عضلانی پیشرونده بازوها و پاها، اختلالات راه رفتن (gait)، مشکلات دفع ادرار، اختلال شناختی خفیف یا زوال عقل و یا سایر ناهنجاریها باشد.
علل بیماری اندرسن
همان طور که در بالا ذکر شد، بیماری اندرسن یک اختلال در متابولیسم گلیکوژن است. متابولیسم به تمام فرآیندهای شیمیایی در بدن، از جمله تجزیه مواد پیچیده به مواد ساده تر و فرآیندهایی که در آن مواد پیچیده از مواد ساده تر ساخته میشود، اشاره دارد. اختلالات متابولیک ناشی از عملکرد غیر طبیعی یک پروتئین یا آنزیم خاص است که فعالیتهای شیمیایی خاصی را در بدن تسریع میکند.
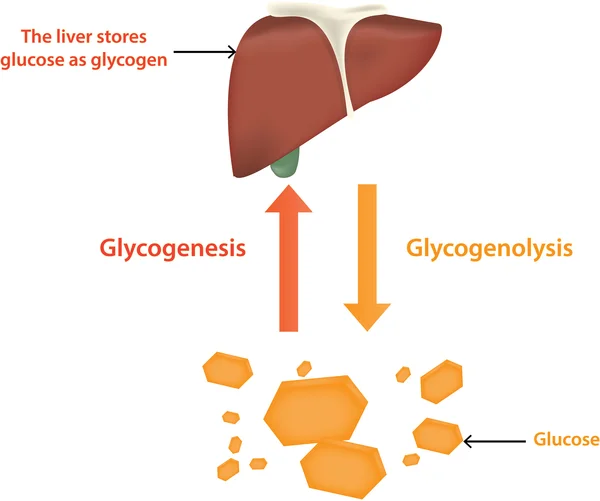
گلیکوژن (Glycogen) اصلی ترین کربوهیدرات ذخیره شده در سلولهای بدن است. این یک کربوهیدرات پیچیده (پلی ساکارید یا polysaccharide) است که از چندین مولکول قند تشکیل شده است که به هم متصل شده اند و یک زنجیره طولانی را تشکیل میدهند. گلیکوژن که عمدتاً در کبد و ماهیچهها ذخیره میشود، به قند ساده (مونوساکارید یا monosaccharide) گلوکز تبدیل میشود و در صورت نیاز وارد جریان خون میشود. هنگامی که سطح قند خون افزایش مییابد، مازاد آن برای ذخیره به گلیکوژن تبدیل میشود. گلوکز منبع اصلی انرژی بدن برای متابولیسم سلولی است.
بیماری اندرسن با کمبود فعالیت آنزیم شاخه ساز گلیکوژن (glycogen-branching enzyme) یا GBE (که معمولاً تعداد نقاط انشعاب را در طول تشکیل گلیکوژن افزایش میدهد) مشخص میشود.
در بیشتر موارد، کمبود فعالیت GBE منجر به تجمع عمومی گلیکوژن غیر طبیعی ساختاری (یعنی با زنجیرههای بیرونی بلند و بدون انشعاب) در بافتهای مختلف بدن میشود. چنین رسوب بافتی در کبد، ماهیچه، سلولهای عصبی، قلب، روده، پوست و غیره نشان داده شده است. بیماری اندرسن گاهی اوقات آمیلوپکتینوز (amylopectinosis) نامیده میشود زیرا گلیکوژن غیر طبیعی از نظر ساختار شبیه به کربوهیدرات پیچیده دیگری است که به نام آمیلوپکتین (amylopectin) شناخته میشود.
جهشهای اختصاصی مختلفی در ژن GBE در افراد مبتلا به بیماری اندرسن، از جمله افراد با فرم کلاسیک کبدی، مبتلایان به بیماری کبدی غیر پیشرونده و نوزادان با فرم شدید عصبی-عضلانی شناسایی شده است. تحقیقات بیشتری برای تعیین اینکه آیا جهشهای خاصی ممکن است با انواع خاصی از بیماری مرتبط باشند، مورد نیاز است.
بیماری اندرسن به عنوان یک صفت اتوزومال مغلوب به ارث میرسد. ویژگیهای انسانی، از جمله بیماریهای ژنتیکی کلاسیک، محصول تعامل دو ژن است که یکی از پدر و دیگری از مادر دریافت میشود.
اختلالات ژنتیکی مغلوب زمانی اتفاق میافتد که یک فرد دو نسخه از یک ژن غیر طبیعی را برای یک صفت به ارث میبرد، یکی از هر والدین. اگر فردی یک ژن طبیعی و یک ژن برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود اما معمولا علائمی از خود نشان نمیدهد. خطر انتقال ژن معیوب و داشتن فرزند مبتلا برای دو والدین ناقل در هر بارداری 25 درصد است.
خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس اینکه کودک از هر دو والدین ژنهای طبیعی دریافت کند و از نظر ژنتیکی برای آن صفت خاص طبیعی باشد 25 درصد است. این خطر برای مردان و زنان یکسان است.
همه افراد حامل 4-5 ژن غیر طبیعی هستند. والدینی که از خویشاوندان نزدیک (فامیلی) هستند، شانس بیشتری نسبت به والدین غیر خویشاوند دارند که هر دو دارای ژن غیرطبیعی مشابهی باشند که خطر بچه دار شدن با اختلال ژنتیکی مغلوب را افزایش میدهد.
جمعیتهای آسیب دیده
همان طور که در بالا ذکر شد، شکل کلاسیک کبدی بیماری اندرسن معمولاً در ماههای اول زندگی آشکار میشود. با این حال، اشکال دیگری از این بیماری نیز توصیف شده است که ممکن است در بدو تولد، در اواخر دوران کودکی یا در بزرگسالی آشکار شود. به نظر میرسد که مردان و زنان به تعداد نسبتاً مساوی تحت تأثیر قرار میگیرند.
فراوانی همه بیماریهای ذخیره گلیکوژن از هر 20000 تا 25000 تولد زنده یک نفر تخمین زده میشود.
با این حال، برخی از محققان پیشنهاد میکنند که فرکانس واقعی ممکن است بیشتر باشد زیرا برخی از افراد مبتلا به اشکال خاصی از بیماری ذخیرهسازی گلیکوژن ممکن است حداقل علائمی داشته باشند که تشخیص داده نشده باقی میمانند.
اختلالات با علائم مشابه
علائم اختلالات زیر ممکن است مشابه علائم بیماری اندرسن باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد:
تعدادی از بیماریها و شرایط دیگر وجود دارد که ممکن است با علائم و نشانههای خاصی مشابه علائم بالقوه مرتبط با بیماری اندرسن مشخص شوند. اینها شامل اختلالات اضافی متابولیسم گلیکوژن (به عنوان مثال، بیماریهای ذخیره گلیکوژن) است. همان طور که در بالا ذکر شد، این بیماریها به دلیل کمبود یک یا چند آنزیم دخیل در تشکیل یا تجزیه گلیکوژن است که منجر به تجمع مقادیر یا اشکال غیر طبیعی گلیکوژن در بافتهای خاص بدن، به ویژه کبد، ماهیچههای اسکلتی و یا میشود. ماهیچه قلب بیشتر این بیماریها به صورت یک صفت اتوزومال مغلوب به ارث میرسند.
تشخیص
بیماری اندرسن معمولاً پس از تولد (پس از تولد یا postnatally) در دوران نوزادی یا کودکی (یا در برخی موارد، بزرگسالی) بر اساس ارزیابی بالینی کامل، شناسایی یافتههای فیزیکی مشخصه، سابقه کامل بیمار و خانواده و نتایج تستهای تخصصی مختلف تشخیص یا تأیید میشود. برداشتن (بیوپسی یا biopsy) و بررسی میکروسکوپی نمونههای کوچک بافتهای خاص (مانند کبد، ماهیچههای اسکلتی، قلب، پوست، عصب محیطی) ممکن است رسوب غیر طبیعی مواد شبه آمیلوپکتین را نشان دهد.
با این حال، آزمایش برای تایید تشخیص بیماری اندرسن نیاز به تشخیص فعالیت ناقص GBE (آزمایش آنزیم غیر مستقیم یا indirect enzyme assay)، مانند در بافت کبد، ماهیچه، سلولهای پوست خاص (فیبروبلاستهای کشت شده یا cultured fibroblasts)، گلبولهای سفید (لکوسیتها یا leukocytes)، گلبولهای قرمز خون (گلبولهای قرمز) و سلولهای عصبی یا سایر بافتها دارد.
گزارشها نشان میدهد که برای افراد مبتلا به بیماری بدن پلی گلوکوزان بالغ، بیوپسی عصب محیطی یا ارزیابی لکوسیتها برای تشخیص مورد نیاز است زیرا فعالیت کمبود GBE محدود به چنین بافتهایی است. علاوه بر این، ممکن است کمبود نسبی GBE (به عنوان مثال در گلبولهای قرمز، لکوسیتها، فیبروبلاستها) در افرادی که حامل یک نسخه از یک ژن جهش یافته برای بیماری اندرسن (ناقلین هتروزیگوت یا heterozygous carriers) هستند، شناسایی شود.
ارزیابی تشخیصی معمولاً شامل مطالعات مختلفی برای کمک به شناسایی و مشخص کردن ناهنجاریهای خاصی است که ممکن است با این اختلال مرتبط باشد. چنین آزمایشاتی ممکن است شامل مطالعات آزمایشگاهی مختلف (به عنوان مثال، شمارش کامل خون، آزمایشات عملکرد کبد، مطالعات قند خون و غیره)،تکنیکهای تصویر برداری تخصصی (مانند سونوگرافی شکم، سی تی اسکن و یا MRI)، آزمایشی که فعالیت الکتریکی عضله اسکلتی را در حالت استراحت و در حین انقباض عضلانی ثبت میکند (الکترومیوگرافی [EMG])، مطالعاتی برای کمک به ارزیابی ساختار و عملکرد قلب، مانند مطالعات اولتراسوند قلب (اکوکاردیوگرافی یا echocardiography) و یا تستهای دیگر باشد.
در برخی موارد، تشخیص بیماری اندرسن قبل از تولد (قبل از تولد) با آزمایشهای تخصصی ممکن است پیشنهاد شود. اینها شامل مطالعاتی است که ممکن است کاهش فعالیت GBE را در سلولهای جنینی خاص که از طریق آمنیوسنتز (amniocentesis) یا نمونه برداری از پرزهای جفتی (CVS یا chorionic villus sampling) به دست آمده اند، شناسایی کند. در طول آمنیوسنتز، نمونهای از مایعی که جنین در حال رشد را احاطه کرده است، برداشته و آنالیز میشود، در حالی که CVS شامل برداشتن نمونههای بافت از بخشی از جفت است. علاوه بر این، در صورت وجود، آنالیز جهش DNA ممکن است در موارد منتخب استفاده شود.
درمان بیماری اندرسن
درمان بیماری اندرسن به سمت علائم خاصی است که در هر فرد آشکار است. چنین درمانی ممکن است به تلاشهای هماهنگ تیمی از متخصصان پزشکی مانند متخصصان اطفال یا متخصصین داخلی، پزشکانی که اختلالات دستگاه گوارش را تشخیص و درمان میکنند، متخصصان مغز و اعصاب، متخصصان قلب و عروق، متخصصان تغذیه و یا سایر متخصصان مراقبتهای بهداشتی نیاز داشته باشد.
درمانهای خاص علامتی و حمایتی هستند و ممکن است شامل مدیریت طولانی مدت سیروز و اختلال در عملکرد کبد، بیماری عصبی عضلانی و یا اختلال عملکرد قلب باشد. درمان معمولاً نیاز به اقدامات رژیمی برای حفظ سطح طبیعی گلوکز در خون (نورموگلیسمی یا normoglycemia) و تأمین مصرف کافی مواد مغذی به منظور بهبود عملکرد کبد و قدرت عضلانی دارد. برای مواردی که کاردیومیوپاتی وجود دارد، مدیریت بیماری توصیه شده ممکن است شامل استفاده از برخی داروها، مانند درمان نارسایی قلبی و بهبود برون ده قلبی مداخله جراحی و یا اقدامات دیگر باشد.
در افراد مبتلا به نارسایی پیشرونده کبد، پیوند کبد (liver transplantation) انجام شده است و ممکن است در برخی موارد موثر باشد. بر اساس گزارشهای موجود در ادبیات پزشکی، پس از پیوند، برخی از بیماران ممکن است به تجمع تدریجی گلیکوژن غیر طبیعی در سایر اندامها مانند قلب مبتلا شوند که منجر به عوارض بالقوه تهدید کننده زندگی میشود.
با این حال، گزارشها نشان میدهد که اکثر بیماران عوارض عصبی-عضلانی یا قلبی نداشته اند (یعنی در طول دورههای پیگیری تا 13 سال). علاوه بر این، در برخی از این بیماران، تجمع گلیکوژن در قلب و عضله اسکلتی پس از پیوند کاهش یافته است. با این حال، کارشناسان توصیه میکنند که اثر بخشی (کارآمدی) طولانی مدت پیوند کبد و تأثیر آن بر سایر سیستمهای اندام در افراد مبتلا به بیماری اندرسن نامشخص است. بنابراین، تحقیقات بیشتری برای تعیین ایمنی و اثر بخشی طولانی مدت پیوند کبد و تأثیر آن بر پیشرفت بیماری در بیماری کلاسیک اندرسن مورد نیاز است.
مشاوره ژنتیک برای افراد مبتلا به بیماری اندرسن و اعضای خانواده مفید خواهد بود. درمان دیگر این اختلال علامتی و حمایتی است.
همچنین بخوانید:
مترجم: فاطمه فریادرس