



خلاصهای از بیماری آکندروژنز
آکندروژنز گروهی از دیسپلازیهای اسکلتی (skeletal dysplasias) نادر است که با کوتاه شدن شدید دستها و پاها در رابطه با تنه، رشد غیر طبیعی دندهها، مهرهها و سایر ناهنجاریهای اسکلتی مشخص میشود. مشکلات سلامتی مرتبط با این شرایط تهدید کننده زندگی هستند و اکثر نوزادان مبتلا مرده به دنیا میآیند یا اندکی پس از تولد به دلیل نارسایی تنفسی میمیرند. همه انواع آکندروژنز عارضههایی ژنتیکی هستند. نوع IA و نوع IB، اختلالات اتوزومال مغلوب هستند، در حالی که آکندروژنز نوع II یک اختلال اتوزومال غالب است. همه انواع آکندروژنز، دیسپلازی اسکلتی بسیار شدید هستند که معمولاً با معاینه اولتراسوند قبل از تولد در اوایل هفته 14-17 سن حاملگی تشخیص داده میشوند.
آکندروژنز چیست؟
اصطلاح آکندروژنز برای اولین بار در ادبیات پزشکی در سال 1952 توسط یک آسیب شناس ایتالیایی به نام مارکو فراکارو (Marco Fraccaro) استفاده شد. آکندروژنز از زبان یونانی گرفته شده است و به معنای “تولید نکردن غضروف” است. آکندروژنز متعلق به گروهی از دیسپلازیهای اسکلتی (همچنین استئوکندرودیسپلازیا یا osteochondrodysplasias نامیده میشود) است که در واقع یک اصطلاح گسترده برای گروهی از اختلالات (حدود 450 تشخیص بالینی) است که با رشد یا توسعه غیر طبیعی غضروف و استخوان مشخص میشوند.
زیر بخشها
- آکندروژنز نوع IA (نوع هیوستون-هریس یا Houston-Harris type)
- آکندروژنز نوع IB (نوع فراکارو یا Fraccaro type)
- آکندروژنز نوع II (نوع لانگر-سالدینو یا Langer-Saldino type)
علائم و نشانهها
آکندروژنز با تولد زودرس، تجمع غیرطبیعی مایع در بدن (هیدروپس فتالیس یا hydrops fetalis) و سر که ممکن است شکل غیر طبیعی داشته باشد و کمتر استخوانی شده باشد، مشخص میشود. سر ممکن است به طور نامتناسبی بزرگ به نظر برسد، زیرا بدن کوچک است. علاوه بر این، افراد مبتلا دارای اندامها و دندههای بسیار کوتاه، گردن کوتاه، مهرههای صاف و بسیاری دیگر از استخوانهای اسکلتی هستند که به درستی رشد نکردهاند.
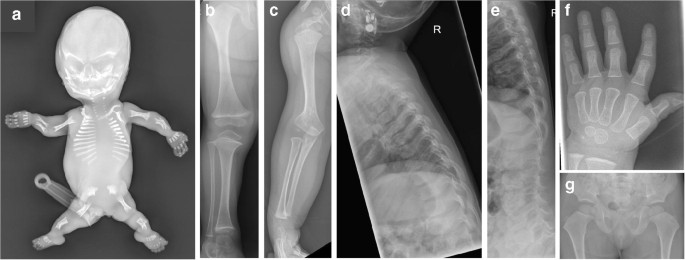
در نوزادانی که با این اختلال متولد میشوند، شکم برجسته و قفس سینه کوچک است. سایر ناهنجاریها شامل بسته شدن ناقص سقف دهان (شکاف کام)، کدر شدن قرنیه و بدشکلی گوش است. این اختلال یا قبل از تولد یا مدت کوتاهی پس از تولد معمولاً به دلیل توسعه نیافته بودن قفسه سینه و داشتن ریههای کوچک، تهدید کننده زندگی است.
آکندروژنز نوع IA (نوع هیوستون-هریس) با ناهنجاریهای مختلف صورت (صورت صاف، چشمهای بیرون زده و زبان بیرون زده یا فقط ناهنجاریهای جزئی صورت)، تنه و اندام کوتاه، دندههای مهره ای کوتاه و استخوانهای نازک جمجمه (کمبود استخوانی شدن جمجمه) مشخص میشود. تشکیل استخوان در ستون فقرات، لگن و اندامها غیرطبیعی است اما درجه شدت درگیری اسکلتی ممکن است متغیر باشد. با این حال، قفسه سینه کوچک منجر به توسعه نیافتگی ریهها و مرگ بلافاصله پس از تولد میشود.
آکندروژنز نوع IB (نوع فراکارو) با تنه و اندام کوتاه، سینه باریک و شکم برجسته مشخص میشود. نوزادان مبتلا ممکن است برآمدگی در اطراف ناف (فتق ناف یا umbilical hernia) یا نزدیک کشاله ران (inguinal hernia) داشته باشند و انگشتان دست و پا کوتاه و پاهایشان به سمت داخل چرخیده باشد. صورت ممکن است صاف باشد، کام ممکن است شکاف داشته باشد و گردن معمولا کوتاه باشد. در برخی موارد، بافت نرم گردن ممکن است به طور غیر طبیعی ضخیم شود.
آکندروژنز نوع IB به عنوان یک اختلال سولفاتاسیون (sulfation disorder)، گروه کوچکی از اختلالات مرتبط با جهش در ژن SLC26A2 طبقه بندی میشود. این گروه شامل دیسپلازی دیاستروفیک (diastrophic dysplasia) و دیسپلازی چندگانه اپی فیزیال مغلوب (recessive multiple epiphyseal dysplasia) است که دارای شرایط خفیف تری هستند. توجه به این نکته مهم است که یک نوع تشخیص به تشخیص دیگر در حین رشد کودک تغییر نمیکند، حتی اگر تغییرات ژنتیکی در همان ژن باشد.
آکندروژنز نوع II (نوع لانگر-سالدینو) با قفسه سینه باریک، استخوانهای کوچک یا غیرعادی کوتاه در بازوها و یا پاها، دندههای نازک، مهرههای صاف یا استخوانبندی ضعیف بدنه مهرهها، ریههای توسعه نیافته، چانه کوچک، شکاف کام و پاهای کلابی (club feet) تشکیل استخوان در ستون فقرات و لگن غیر طبیعی است. تجمع غیر طبیعی مایع ممکن است رخ دهد (هیدروپس فتالیس) و شکم بزرگ شود.
علل
هر نوع آکندروژنز به دلیل جهش در یک ژن خاص ایجاد میشود. ژنها دستورالعملهایی را برای ایجاد پروتئینهایی ارائه میدهند که نقش مهمی در بسیاری از عملکردهای بدن دارند. هنگامی که یک جهش در یک ژن رخ میدهد، محصول پروتئینی ممکن است معیوب یا ناکارآمد بوده یا أصلا وجود نداشته باشد. بسته به عملکرد پروتئین خاص، این میتواند بر بسیاری از اندامهای بدن تأثیر بگذارد.
جهشهای ژنی که باعث آکندروژنز نوع IA و نوع IB میشوند به روش اتوزومال مغلوب به ارث میرسند. اختلالات ژنتیکی مغلوب زمانی اتفاق میافتد که یک فرد دو نسخه از یک ژن غیرطبیعی را برای یک صفت به ارث میبرد، یکی از هر یک از والدین. اگر فردی یک ژن طبیعی و یک ژن را برای بیماری دریافت کند، فرد ناقل بیماری است اما معمولاً علائمی از خود نشان نمیدهد. خطر داشتن فرزند مبتلا برای دو والدین ناقل در هر بارداری 25 درصد است.
خطر داشتن فرزندی که مانند والدین ناقل است در هر بارداری 50 درصد است. شانس دریافت ژنهای طبیعی از هر دو والدین برای کودک 25 درصد است. این خطر برای مردان و زنان یکسان است.
همه افراد حامل چندین ژن غیر طبیعی هستند. والدینی که از خویشاوندان نزدیک (فامیلی) هستند، شانس بیشتری نسبت به والدین غیر فامیلی دارند که ژن غیرطبیعی مشابهی داشته باشند که خطر بچه دار شدن با یک اختلال ژنتیکی مغلوب نادر را افزایش میدهد.
آکندروژنز نوع IA در اثر جهش در ژن TRIP11 ایجاد میشود. آکندروژنز نوع IB در اثر جهش در ژن SLC26A2 ایجاد میشود. این دو ژن برای انتقال کارآمد سلولی پروتئینهای غضروفی خاص مورد نیاز برای ساخت اسکلت و سایر بافتها مورد نیاز هستند. جهش ژن TRIP11 منجر به کمبود پروتئین 210 مرتبط با میکروتوبول گلژی (Golgi microtubule associated protein 210) میشود. این پروتئین در اکثر انواع سلولهای بدن یافت میشود.
محصول پروتئینی ژن SLC26A2 یک ناقل سولفات است که در رشد و عملکرد صحیح غضروف نقش دارد. غضروف بافت تخصصی است که به عنوان بافر یا بالشتک در مفاصل عمل میکند. بیشتر اسکلت جنین از غضروف تشکیل شده است که به آرامی به استخوان تبدیل میشود.
جهش ژنی که باعث آکندروژنز نوع II میشود به دلیل تغییر اتوزومال غالب در ژن COL2A1 ایجاد میشود. اختلالات ژنتیکی غالب زمانی اتفاق میافتد که تنها یک نسخه از یک ژن غیرطبیعی برای ایجاد یک بیماری خاص ضروری باشد. اکثر موارد آکندروژنز نوع II ناشی از یک جهش جدید (de novo) در ژن COL2A1 است، به این معنی که خطر ابتلا والدین یک نوزاد مبتلا به کودک دیگری با همان بیماری بیشتر از 1 درصد نیست. این ژن کلاژن نوع II را کد میکند.
کلاژن یکی از فراوانترین پروتئینها در بدن و بلوک ساختمانی اصلی بافت همبند است که ماده بین سلولهای بدن است که به بافت، شکل و استحکام میدهد. انواع مختلفی از کلاژن وجود دارد که با اعداد رومی مشخص میشوند. کلاژن نوع II بیشتر در غضروف و مایع ژله مانندی که مرکز چشم را پر میکند (vitreous) است. کلاژن در استخوان نیز یافت میشود.
موارد بسیار نادری وجود دارد که خواهر و برادر نوزادان مبتلا به آکندروژنز نوع II تحت تأثیر قرار گرفته باشند. این به احتمال زیاد به دلیل وجود بیش از یک رده سلولی در تخمک یا اسپرم از والدین است (موزاییسم سلول جنسی یا germline mosaicism). در موزائیسم ژرمینال، برخی از سلولهای تولید مثلی والدین (سلولهای زاینده) حامل جهش ژن COL2A1 هستند، در حالی که سایر سلولهای زایا حاوی ژنهای طبیعی COL2A1 (موزاییکیسم یا mosaicism) هستند. سایر سلولهای بدن والدین دارای جهش نیستند، بنابراین این والدین تحت تأثیر قرار نمیگیرند.
در نتیجه، یک یا چند فرزند والدین ممکن است جهش COL2A1 ژن سلول زایا را به ارث ببرند که منجر به ایجاد آکندروژنز II میشود، در حالی که والدین این اختلال را ندارند (ناقل بدون علامت). زمانی که والدینی که ظاهراً مبتلا نیستند، بیش از یک فرزند با شرایط ژنتیکی اتوزومال غالب یکسان داشته باشند، ممکن است مشکوک به موزائیسم سلول جنسی باشد.
احتمال انتقال جهش زایا موزاییکی توسط والدین به کودک بستگی به درصد سلولهای زایای والدین دارد که دارای جهش هستند در مقابل درصدی که این جهش را ندارند. هیچ آزمایشی برای جهش ژرمینال قبل از بارداری وجود ندارد. آزمایش در اوایل بارداری ممکن است در دسترس باشد و بهتر است مستقیماً با یک متخصص ژنتیک صحبت شود.
جمعیتهای آسیب دیده
آکندروژنز در مردان و زنان به تعداد مساوی تأثیر میگذارد. آکندروژنز نوع IA و نوع IB اختلالات بسیار نادری هستند و شیوع آنها ناشناخته است. آکندروژنز نوع II تقریباً در یک نوزاد در هر 40000-60000 مورد تولد رخ میدهد.
اختلالات با علائم مشابه
دیسپلازیهای اسکلتی (استئوکندرودیسپلازیها) یک اصطلاح کلی برای گروهی از اختلالات است که با رشد یا تکامل غیر طبیعی غضروف و یا استخوان مشخص میشود. برخی از اشکال در مدت کوتاهی پس از تولد باعث عوارض تهدید کننده زندگی میشوند، در حالی که برخی دیگر ممکن است عوارض تهدید کننده زندگی ایجاد کنند یا نکنند.
برخی از اشکال در اوایل زندگی عوارض تهدید کننده زندگی ایجاد نمیکنند. دیسپلازیهای اسکلتی میتواند با کوتاهی قد یا کوتاه شدن تنه و اندامها همراه باشد. بسته به اختلال خاص ممکن است ناهنجاریهای اضافی مختلفی وجود داشته باشد. تقریباً 450 نوع دیسپلازی اسکلتی با بیش از 360 ژن ایجاد کننده وجود دارد. در زیر چندین فرم مورد بحث قرار گرفته است. تمایز بین آنها با روشهای رادیوگرافی و مولکولی انجام میشود.
دیسپلازی Kniest (Kniest dysplasia) یکی از چندین اشکال دیسپلازی اسکلتی است که در اثر تغییر (جهش) در ژن COL2A1 ایجاد میشود. این ژن در تولید پروتئین خاصی دخیل است که کلاژن نوع II را تشکیل میدهد که برای رشد طبیعی استخوانها و سایر بافت همبند ضروری است. تغییرات در ترکیب کلاژن نوع II منجر به رشد غیر طبیعی اسکلتی و در نتیجه انواع بیماریهای اسکلتی مادرزادی به نام دیسپلازی اسکلتی میشود. برخی از علائم و نشانههای دیسپلازی Kniest، مانند کوتاهی قد، بزرگ شدن زانوها و شکاف کام معمولاً در بدو تولد وجود دارند. علائم دیگر با افزایش سن ایجاد میشود.
سندرم کموملیک (Campomelic syndrome) یک اختلال مادرزادی نادر است که در آن ناهنجاریهای متعددی وجود دارد. این بیماری در اثر جهش در ژن SOX9 ایجاد میشود و با شکل خمیده و زاویهدار استخوانهای بلند پاها، بهویژه استخوان ساق، مشخص میشود. ناهنجاریهای جزئی متعدد صورت، شکاف کام، سایر ناهنجاریهای اسکلتی مانند ناهنجاریهای ناحیه شانه و لگن و یازده جفت دنده به جای دوازده جفت معمول نیز وجود دارد. توسعه نیافتگی نای، تأخیر رشد در برخی موارد و رشد ناقص اندام تناسلی در مردان به گونه ای که به نظر میرسد زن هستند نیز از دیگر موارد هستند.
هیپوفسفاتازی (Hypophosphatasia) یک اختلال متابولیک ذاتی استخوان است که با نقایص اسکلتی شبیه به راشیتیسم مشخص میشود. علائم ناشی از شکست مواد معدنی استخوان در ته نشین شدن در استخوان جوان و بدون کلسیفیه (استوئید یا osteoid) و در غضروف انتهای استخوانهای بلند (اپی فیز یا epiphyses) در سالهای اولیه است. فعالیت آنزیم آلکالین فسفاتاز در سرم خون و سلولهای استخوانی کمتر از حد طبیعی است. دفع ادرار و غلظت پلاسمایی فسفواتانول آمین و پیروفسفات معدنی به طور غیر طبیعی بالاست. Hypophosphatasia به درمان با Strensig پاسخ میدهد.
دیسپلازی تاناتوفوریک (Thanatophoric dysplasia) یکی از رایج ترین اشکال دیسپلازی اسکلتی کشنده است. با سر بزرگ شده، استخوانهای کوتاه و در نهایت کماندار در بازوها و پاها، دندههای کوچک و کوتاه، قفسه سینه باریک و مهرههای صاف مشخص میشود. مقدار غیر طبیعی مایع آمنیوتیک زیادی وجود دارد. دیسپلازی Thanatophoric در اثر جهش در ژن FGFR3 ایجاد میشود.
سندرم دنده کوتاه-پلی داکتیلی (Short-rib-polydactyly syndrome) شامل انواع مختلفی از دیسپلازی اسکلتی است. نوزاد ممکن است دارای شکاف لب و کام، تغییر شکل گوش و قفسه سینه باریک با دندههای کوتاه باشد. کلیهها و همچنین اندامهای جنسی اغلب تغییر شکل میدهند (کیستیک یا cystic). ممکن است ناهنجاریهای مغزی و عدم وجود کیسه صفرا وجود داشته باشد. این اختلال اغلب در نتیجه رشد ناکافی ریهها تهدید کننده زندگی است.
تشخیص
آکندروژنز با ویژگیهای فیزیکی، یافتههای اشعه ایکس (رادیوگرافی یا radiographic) و بررسی نمونههای بافت زیر میکروسکوپ (هیستولوژی یا histology) تشخیص داده میشود. آزمایشات ژنتیکی مولکولی برای جهش در ژن SLC26A2 میتواند برای تایید تشخیص آکندروژنز نوع B۱ استفاده شود.
تشخیص پیش از تولد آکندروژنز با سونوگرافی پس از 14-15 هفته بارداری امکان پذیر است. تشخیص قبل از تولد با نمونه برداری از پرزهای کوریونی (هفته 12-10 بارداری) یا آمنیوسنتز (هفته 15-18 بارداری) در صورتی امکان پذیر است که جهشهای ژنی خاص در یکی از اعضای خانواده شناسایی شده باشد.
درمان
درمان آکندروژنز علامتی (symptomatic) و حمایتی (supportive) است و شامل مراقبتهای تسکینی (palliative care) است که در آن پزشکان تلاش میکنند تا درد، استرس و علائم خاص مرتبط با این اختلال را کاهش یا به حداقل برسانند. مشاوره ژنتیک برای خانوادههایی که کودک مبتلا دارند توصیه میشود. حمایت روانی اجتماعی برای کل خانواده نیز ضروری است.
همچنین بخوانید:
مترجم: فاطمه فریادرس