


دستهبندی نشده
آنالیز مرحله دوم (Secondary Analysis) داده های NGS شامل و Alignment آن با ژنوم مرجع
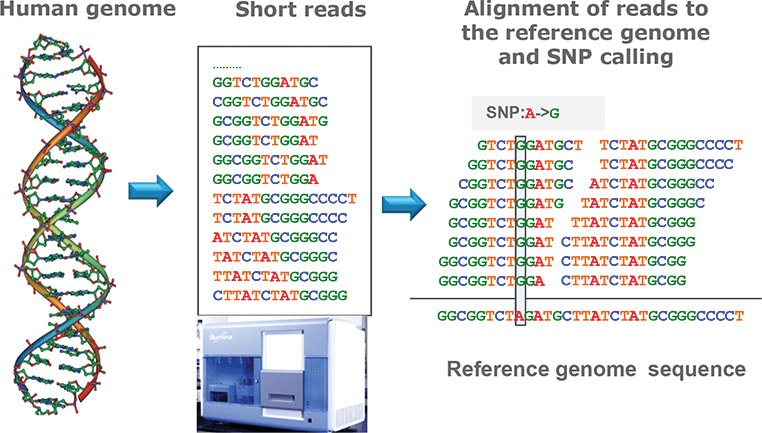
آنالیز داده ها در سطح دوم شامل هم ردیف سازی توالی خوانشی کنترل کیفی شده در برابر ژنوم مرجع می باشد تا در نهایت در یک فایل واریانت ها فراخوانی شوند (Variant Calling).
تراز کردن، که به آن mapping نیز گفته می شود، یک مرحله ضروری در توالی یابی مجدد re-sequencing است. با توالی یابی یک گونه از یک ارگانیسم از قبل، و ایجاد یک توالی مرجع، توالی یابی مجدد ارگانیسم های بیشتری از همان گونه به ما این امکان را می دهد تا با مقایسه سکانس توالی یابی شده جدید با سکانس مرجع به تفاوت های ژنتیکی بین آن دو پی ببریم.
هم ترازی داده ها یک روش نسبتاً ساده برای تشخیص تنوع در نمونه ها است. موارد خاصی وجود دارد (مانند ژنهای جدید در نمونه توالییابی شده که در توالی مرجع موجود یافت نمیشوند) که نمیتوان آنها را به تنهایی با همترازی شناسایی کرد. در حالی که روشهای دیگر، مانند مونتاژ de novo، به طور بالقوه قویتر هستند، اما دستیابی به آنها با روشهای توالییابی فعلی بسیار سختتر یا برای برخی ارگانیسمها غیرممکن است.
توالییابی نسل بعدی عموماً خوانش های کوتاه کوتاه کمتر از 200 باز ایجاد میکند (در مقایسه با خوانشهای به نسبت طولانی توالی یابی سنگرمعادل حدودا 1000 باز). برای مقایسه DNA نمونه توالییابی شده با توالی مرجع آن، باید بخش مربوط به آن توالی را در دادههای توالییابی خود پیدا کنیم. به این کار Alignment یا هم تراز کردن در برابر توالی مرجع گفته می شود. پس از انجام این کار، میتوان پی به تنوعات و یا همان واریانت ها (به عنوان مثال SNP) در نمونه برد که در این میان یک سری مشکلات ایجاد خواهد شد از جمله:
- خوانش های کوتاه با اطلاعات موقعیتی بازها همراه نیستند، یعنی نمی دانیم از چه بخشی از ژنوم آمده اند.
- توالی مرجع می تواند بسیار طولانی باشد (حدود 3 میلیارد باز برای انسان)، که یافتن یک منطقه منطبق با توالی ها را بسیار سخت می کند.
- همچنین چون خوانش های ما کوتاه هستند، ممکن به چندین منطقه به ژنوم Align شوند که این اتفاق به خصوص در مناطق تکراری رخ می دهد.
- همینطور اگر ما فقط به دنبال مطابقت کامل با ژنوم مرجع بودیم، هرگز تغییری نمی دیدیم. بنابراین، ما باید اجازه دهیم برخی ناهماهنگی ها و تغییرات ساختاری کوچک (InDels) در خوانش هایمان وجود داشته باشد.
- هر تکنولوژی توالی یابی می تواند با خطا همراه باشد. هم زمان با تغییرات “real”، می بایست سطح پایینی از خطاهای ترتیبی را در خوانش ها تحمل کنیم و در ادامه آنها را از تغییرات و یا واریانت های “real” جدا کنیم.
همه این کارها می بایست برای هر یک از میلیون ها خوانش در داده های حاصل از توالی یابی خ انجام شود.
برای هر یک از خوانش های کوتاه در فایل FASTQ، یا یک مکان در دنباله مرجع می بایست تعیین شود و یا این که هیچ مکانی برای هم ترازی مشاهده نشود. این هدف با مقایسه سکانس خوانده شده با سکانس مرجع به دست می آید. یک الگوریتم هم ترازی سعی می کند تنها یک مکان (ترجیحا منحصر به فرد) را در دنباله مرجع که با خوانش ها مطابقت دارد پیدا کند، در حالی که مقدار معینی عدم تطابق را تحمل می کند تا امکان تشخیص تغییرات بعدی را فراهم کند. تراز یک توالی سکانس شده با یک ژنوم مرجع به شکل زیر خواهد بود:
GCTGATGTGCCGCCTCTCACTTCGGTGGTGAGGTG دنباله مرجع
CTGATGTGCCGCCTCACTTCGGTGGT خوانش کوتاه 1
TGATGTGCCGCCTCTCACTACGGTGGTG خوانش کوتاه 2
GATGTGCCGCCTCACTTCGGTGGTGA خوانش کوتاه 3
GCTGATGTGCCGCCTCTCACTACGGTG خوانش کوتاه 4
GCTGATGTGCCGCCTCTCACTACGGTG خوانش کوتاه 5
در حالی که دو مورد از خوانش ها مطابقت کاملی با مرجع دارند، سه خوانش دیگر هر کدام یک عدم تطابق را نشان میدهند که با رنگ قرمز مشخص شده است (“A” در خوانش، به جای “T” در مرجع). از آنجایی که چندین خوانش وجود دارد که عدم تطابق را نشان می دهد، می توان نتیجه گرفت که این یک تفاوت ژنتیکی واقعی (جهش نقطه ای یا SNP)، به جای یک خطای توالی یابی می باشد.
چندین منبع بالقوه برای خطا در یک پروسه Alignment وجود دارد، از جمله:
آرتیفکت های واکنش زنجیره ای پلیمراز. بسیاری از روش های NGS شامل یک یا چند مرحله PCR هستند. خطاهای PCR به صورت عدم تطابق در تراز نشان داده می شوند و به خصوص خطاها در دورهای اولیه PCR در خوانش های متعدد نشان داده می شوند که به اشتباه نشان دهنده تنوع ژنتیکی در نمونه است. یک خطای مرتبط با تکراری بودن PCR است، که در آن خوانش های یکسان چندین بار اتفاق میافتد و محاسبات coverage را در تراز کردن تغییر میدهند.
خطاهای دستگاه توالی یاب نیز شامل دلایل فیزیکی (مانند روغن روی اسلاید ایلومینا) یا دلایلی همچون خواص DNA توالی یابی شده (مانند هموپلیمرها) خطا ایجاد کند.
سلام،
عالی بود.