



آمیلوئیدوز چیست؟
آمیلوئیدوز (Amyloidosis) یک اختلال سیستمیک است که بر اساس پروتئین پیش ساز به چند نوع طبقه بندی میشود. انواع مختلف آمیلوئیدوز به عنوان سیستمیک (systemic) یا موضعی (localized) طبقه بندی میشوند. آمیلوئیدوز AL (زنجیره سبک ایمونوگلوبولین یا immunoglobulin light chain که از لحاظ تاریخی به عنوان اولیه شناخته میشود) شایع ترین نوع آمیلوئیدوز سیستمیک است. آمیلوئیدوز AL ناشی از ناهنجاری (دیسکرازی یا dyscrasia) نوعی از گلبولهای سفید خون به نام سلولهای پلاسما (plasma cells) در مغز استخوان است و ارتباط نزدیکی با مولتیپل میلوما (multiple myeloma) دارد.
آمیلوئیدوز AA (از لحاظ تاریخی به عنوان ثانویه شناخته میشود) از پروتئین التهابی سرم آمیلوئید A (amyloid A) مشتق شده است. آمیلوئیدوز AA همراه با بیماریهای التهابی مزمن مانند بیماریهای روماتیسمی (rheumatic diseases)، تب مدیترانه ای خانوادگی (familial Mediterranean fever)، بیماری التهابی مزمن روده (chronic inflammatory bowel disease)، سل (tuberculosis) یا آمپیم (empyema) رخ میدهد. آمیلوئیدوز ارثی (Hereditary amyloidosis) یک نوع نادر از آمیلوئیدوز است که توسط یک ژن غیر طبیعی ایجاد میشود.
چندین ژن غیر طبیعی وجود دارد که میتواند باعث آمیلوئیدوز ارثی شود اما شایعترین نوع ارثی ATTR نام دارد و در اثر تغییرات (واریانتهای بیماریزا یا جهش) در ژن ترانس تیرتین (TTR یا transthyretin) ایجاد میشود. آمیلوئیدوز مرتبط با سن که در آن آمیلوئید از نوع وحشی (طبیعی) ترانس تیرتین مشتق میشود یک بیماری به آرامی پیشرونده است که بر قلب افراد مسن، معمولاً مردان و کمتر زنان تأثیر میگذارد و به آن آمیلوئیدوز ATTRwt میگویند.
رسوبات آمیلوئید ممکن است گهگاه به صورت مجزا و بدون شواهدی از بیماری سیستمیک ایجاد شوند. مثانه ایزوله (isolated bladder) یا آمیلوئیدوز تراشه ای (tracheal amyloidosis) شایع ترین تظاهرات این گونه هستند. آمیلوئیدوز بتا 2 میکروگلوبولین مربوط به دیالیز نوعی آمیلوئیدوز سیستمیک است که میتواند در افرادی که دیالیز طولانی مدت کلیه را تجربه کرده اند برای حذف ناخالصیها یا ضایعات انباشته شده در خون با فیلتراسیون مکانیکی رخ دهد.
این شکل از آمیلوئیدوز، همچنین به عنوان AB2MG (آمیلوئید مرتبط با پروتئین بتا-2m) شناخته میشود، با تجمع بتا-میکروگلوبولین (beta2-microglobulin) – نوعی پروتئین آمیلوئید که توسط کلیه با عملکرد طبیعی پاک میشود – مرتبط است. آمیلوئیدوز بتا2-میکروگلوبولین مربوط به دیالیز در بیماران مبتلا به بیماری کلیوی نزدیک به مرحله نهایی رخ میدهد. این دارو بر افرادی با عملکرد کلیوی طبیعی یا خفیف کاهش یافته یا بیمارانی که پیوند کلیه دارند تأثیر نمیگذارد.
زیر بخشها
- آمیلوئیدوز AL
- آمیلوئیدوز ATTR – آمیلوئیدوز ATTRv یا ATTRwt
- آمیلوئیدوز AA
- آمیلوئیدوز بتا2 میکروگلوبولین مرتبط با دیالیز (dialysis-related beta2-microglobulin amyloidosis یا AB2MG)
علائم و نشانهها
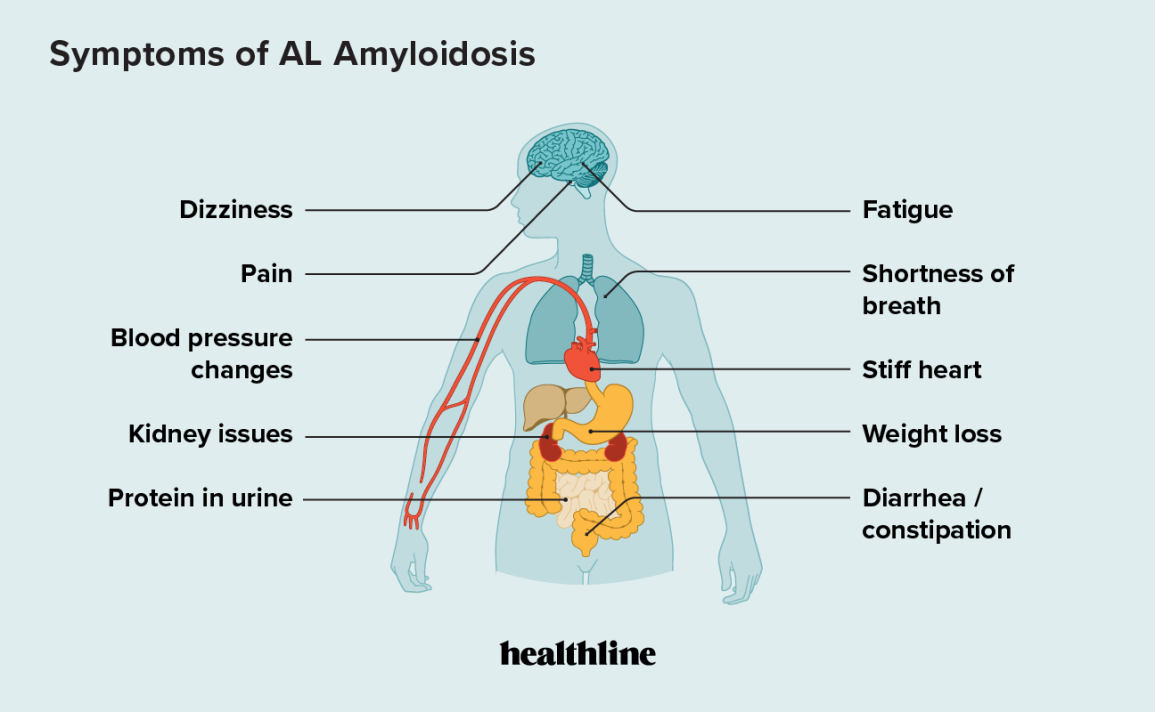
آمیلوئیدوز معمولا یک بیماری چند سیستمی است که منجر به طیف وسیعی از تظاهرات بالینی میشود. در نتیجه، یک بیمار ممکن است به یکی از چندین متخصص و فوق تخصص، معمولاً یک نفرولوژیست، متخصص قلب و اعصاب مراجعه کند یا به او ارجاع داده شود. پیشرفتهای اخیر در درمان، تشخیص زودهنگام و دقیق را در صورتی که بیمار به طور کامل از آن بهره مند شود، حیاتی کرده است. اکثر بیماران بیش از یک عضو درگیر دارند و بنابراین یافتن ترکیبی از هر یک از ویژگیهای زیر باید شک آمیلوئیدوز را تشدید کند:
کلیه اندامی است که بیشتر درگیر در آمیلوئیدوز AL و آمیلوئیدوز AA است، با این حال، به ندرت در آمیلوئیدوز ارثی (خانوادگی) درگیر است. مقدار بیش از حد پروتئین در ادرار (پروتئینوری یا proteinuria) تظاهرات معمول درگیری کلیه است و معمولاً سنگین است و منجر به سندرم نفروتیک (nephrotic syndrome) میشود.
به ندرت، این بیماری باعث افزایش بیش از حد اوره و سایر مواد زائد نیتروژنی در خون (آزوتمی پیشرونده ییا progressive azotemia) به عنوان تظاهرات اولیه بیماری کلیوی میشود. تجمع غیر طبیعی مایعات مانند تورم پاها و شکم، در غیاب نارسایی قلبی از ویژگیهای سندرم نفروتیک است، همچنین وجود کلسترول اضافی در خون (هیپرکلسترولمی یا hypercholesterolemia) که ممکن است عمیق باشد. کلیهها اغلب کوچک، رنگ پریده و سفت میشوند اما در آمیلوئیدوز، کلیههای بزرگ نیز معمولا دیده میشوند.
این بیماری اغلب قلب را درگیر میکند. قلب معمولاً در آمیلوئیدوز AL و ATTRv درگیر است و شایع ترین فنوتیپ آمیلوئیدوز ATTRwt است. انفیلتراسیون آمیلوئید (Amyloid infiltration) قلب منجر به ضخیم شدن دیواره بطن و ایجاد نارسایی قلبی میشود. نارسایی احتقانی قلب به سرعت پیشرونده با دیوارههای بطنی ضخیم، تظاهرات کلاسیک آمیلوئیدوز قلبی AL است. قلب همواره در آمیلوئیدوز ATTRwt، اغلب در آمیلوئیدوز ATTR و به ندرت در آمیلوئیدوز AA درگیر است.
علائم شایع درگیری قلب عبارتند از:
- بزرگ شدن قلب (کاردیومگالی یا cardiomegaly)
- ضربان قلب نامنظم (آریتمی یا arrhythmias)
- ناهنجاریهای قلب که در نوار قلب دیده میشود
نارسایی احتقانی قلب شایع ترین عارضه قلبی آمیلوئیدوز است. رسوبات ندولار (Nodular deposits) آمیلوئید ممکن است روی کیسه غشایی که قلب را احاطه کرده است (پریکارد یا pericardium) و روی پوشش حفرههای قلب یا دریچههای قلب (اندوکارد یا endocardium) وجود داشته باشد.
اگر چه این مورد کمتر از درگیری کلیوی یا قلبی شایع است، نوروپاتی (neuropathy) ممکن است یک مشکل مهم در آمیلوئیدوز باشد. گاهی اوقات، این ویژگی بارز و غالب آمیلوئیدوز AL است.
در انواع خاص آمیلوئیدوز ارثی (به ویژه V30M ATTR که در اصل به عنوان پلی نوروپاتی آمیلوئید خانوادگی یا familial amyloid polyneuropathy شناخته میشود)، ویژگی اصلی بیماری است. نوروپاتی اغلب ماهیتی دردناک و حسی-حرکتی دارد، اگرچه درد نوروپاتی ممکن است گاهی قابل توجه باشد.
این علائم ممکن است شامل نوروپاتی حسی همراه با بی حسی و احساس سوزن سوزن شدن در پاها باشد که به سمت پاها و در نهایت اندامهای فوقانی پیشرفت میکند. نوروپاتی حرکتی (motor neuropathy) با از دست دادن حرکت از پا شروع میشود و به سمت بالا گسترش مییابد.
سندرم تونل کارپال (Carpal tunnel syndrome) معمولاً به دلیل درگیری مستقیم عصب نیست، بلکه به دلیل انفیلتراسیون بافت نرم (soft tissue infiltration) است که باعث فشردگی عصب میانی میشود.
در آمیلوئیدوز ATTRv، نوروپاتی محیطی (peripheral neuropathy) اغلب با نوروپاتی اتونومیک (autonomic neuropathy) همراه است که با یبوست و کاهش میزان تولید عرق (هیپوهیدروز یا hypohidrosis)، افت ناگهانی فشار خون در هنگام ایستادن بیمار (هیپوتانسیون وضعیتی یا postural hypotension) و اختلال در نعوظ در مرد و افت فشار خون – وضعیتی ممکن است عمیق باشد و منجر به غش مکرر (سنکوپال یا syncopal) شود – همراه است. آمیلوئیدوز سیستمیک سیستم عصبی مرکزی را درگیر نمیکند و ارتباطی با بیماری آلزایمر ندارد.
این بیماری ممکن است بر کبد و طحال تاثیر بگذارد. درگیری آمیلوئید در طحال خطر پارگی خود به خودی آن عضو را افزایش میدهد. درجاتی از درگیری کبد در آمیلوئیدوز AL شایع است. این وضعیت همچنین در آمیلوئیدوز AA شایع است اما در آمیلوئیدوز ATTRv دیده نمیشود. در اکثر بیماران درگیری کبدی بدون علامت است.
بزرگ شدن کبد (هپاتومگالی یا hepatomegaly) و بزرگ شدن طحال (سپلنومگالی یا splenomegaly) بارزترین علائم هستند. به طور کلی، کبد انفیلتر شده با آمیلوئید بسیار سخت است و افزایش آنزیمهای کبدی (به ویژه آلکالین فسفاتاز یا alkaline phosphatase) و سایر ناهنجاریهای عملکرد کبدی ممکن است زود تشخیص داده شوند.
به طور کلی، عملکرد کبد تا اواخر دوره بیماری به طور قابل توجهی تحت تأثیر قرار نمیگیرد. افزایش بیلی روبین یک علامت بد است و ممکن است نشان دهنده نارسایی کبدی (hepatic failure) باشد. آمیلوئیدوز کبدی به ندرت به صورت مجزا رخ میدهد و معمولاً با درگیری اندام در جاهای دیگر همراه است.
آمیلوئیدوز همچنین ممکن است بر سیستم گوارشی (هضم کننده یا digestive) تأثیر بگذارد. تجمع آمیلوئید در دستگاه گوارش ممکن است باعث عدم حرکت (motility) در مری و روده کوچک و بزرگ شود. سوء جذب، زخم، خونریزی، فعالیت ضعیف معده، انسداد کاذب دستگاه گوارش، از دست دادن پروتئین و اسهال نیز ممکن است رخ دهد.
از دست دادن چشایی و مشکل در خوردن غذاهای جامد به دلیل بزرگ شدن زبان (ماکروگلوسیا یا macroglossia) در اثر نفوذ آمیلوئید، ممکن است به کاهش وزن کمک کند یا کاهش وزن ممکن است تظاهرات غیر اختصاصی بیماری سیستمیک باشد. در بیماران مبتلا به نوروپاتی اتونومیک، تخلیه معده مختل میشود و در نتیجه احساس سیری زودرس ایجاد میشود.
پوست اغلب در آمیلوئیدوز اولیه درگیر است. درگیری پوستی تقریباً منحصراً به آمیلوئیدوز AL محدود میشود و شامل بافت نرم، پوست و ناهنجاریهای عروقی است. پورپورای دور چشمی (Periorbital purpura) نتیجه شکنندگی مویرگها است و ممکن است پس از سرفه، عطسه یا زور زدن برای اجابت مزاج ظاهر شود.
گاها، ضایعات پورپوریک ممکن است پس از اقدامات ساده ای مانند مالیدن پلکها ایجاد شوند. نفوذ بافت نرم ممکن است باعث ماکروگلوسیا و گرفتگی صدا شود، اگرچه معاینه تارهای صوتی ممکن است طبیعی به نظر برسد. ضایعات پوستی ممکن است قابل مشاهده باشند یا آن قدر کوچک باشند که فقط با میکروسکوپ قابل مشاهده باشند.
ضایعات پاپولار مومی شکل (Waxy looking papular lesions) ممکن است روی صورت و گردن ظاهر شوند. آنها همچنین ممکن است در زیر بازوها (ناحیه زیر بغل)، نزدیک مقعد و کشاله ران ایجاد شوند. نواحی دیگری که ممکن است تحت تأثیر قرار گیرند، نواحی مخاطی مانند کانال گوش یا زبان هستند. نواحی متورم، خونریزی زیر پوست (پورپورا یا purpura)، ریزش مو (آلوپسی یا alopecia)، التهاب زبان (گلوسیت یا glossitis) و خشکی دهان (xerostomia) نیز ممکن است وجود داشته باشد.
مشکلات سیستم تنفسی که با آمیلوئیدوز همراه است اغلب با علائم قلبی موازی است. در شکل موضعی آمیلوئیدوز، گذرگاهها و مجاری هوا ممکن است توسط رسوبات آمیلوئید در سینوسهای بینی، جعبه صدا (حنجره) و گلو (نای) و سیستم نایژکی مسدود شوند. جمع آوری مایعات در فضای پلور (افیوژن پلورال یا pleural effusion) در بیماران مبتلا به نارسایی احتقانی قلب به دلیل آمیلوئیدوز بسیار شایع است اما افیوژنهای مکرر پلورال بزرگ نامتناسب با درجه نارسایی قلبی، آمیلوئیدوز پلورال (pleural amyloidosis) را نشان میدهد.
ناهنجاریهای مفصلی (آرتروپاتی یا arthropathy) در آمیلوئیدوز به دلیل تجمع رسوبات آمیلوئید در پوشش مفاصل (غشاء سینوویال یا synovial membranes) رخ میدهد. این در آمیلوئیدوز AL و گاهی در آمیلوئیدوز مرتبط با دیالیز رخ میدهد. غضروف مفصلی یا غشای سینوویال و مایع نیز ممکن است درگیر شوند. علائم مشابه آرتریت روماتوئید است. رسوبات آمیلوئید در بافت عضلانی ممکن است باعث ضعف عضلانی و تغییرات عضلانی شود (شبهدویوپاتی یا pseudomyopathy). علائم آمیلوئیدوز نیز ممکن است با اختلالات خونریزی آشکار شود.
اینها ممکن است ناشی از کمبود برخی فاکتورهای انعقادی یا رسوبات کوچک آمیلوئید در رگهای خونی داخل پوست باشد.
آمیلوئیدوز بتا 2 میکروگلوبولین مربوط به دیالیز معمولاً استخوانها و مفاصل را تحت تأثیر قرار میدهد. علائم اولیه شامل سندرم تونل کارپال، درد شانه و التهاب غلاف تاندون دست است. گزارشهای موردی از فشار خون شدید ریوی و نارسایی قلبی نیز وجود دارد.
علل
آمیلوئیدوز ناشی از تاخوردگی غیر طبیعی پروتئینهای محلول طبیعی است که منجر به تشکیل فیبریل در یک یا چند اندام، سیستم یا بافت نرم بدن میشود. این تودههای پروتئینی رسوبات آمیلوئید (amyloid deposits) نامیده میشوند و تجمع رسوبات آمیلوئیدی باعث اختلال پیش رونده و نارسایی نهایی اندام آسیب دیده میشود. به طور معمول، پروتئینها تقریباً با همان سرعتی که تولید میشوند، تجزیه میشوند اما این رسوبات آمیلوئیدی پایدار غیر معمول سریعتر از شکسته شدنشان رسوب میکنند.
علت آمیلوئیدوز AL معمولا یک دیسکرازی سلولهای پلاسما (plasma cell dyscrasia) است، یک ناهنجاری اکتسابی از سلولهای پلاسما در مغز استخوان با تولید یک پروتئین غیر طبیعی زنجیره سبک (بخشی از یک آنتی بادی). معمولاً مقدار اضافی پروتئین آنتی بادی تولید میشود و بخش غیر طبیعی زنجیره سبک یا کل مولکول آنتی بادی به شکل رسوبات آمیلوئید در بافتهای بدن تجمع مییابد.
آمیلوئیدوز AA ناشی از فرآیند بیماری التهابی است که بخشی از بیماری زمینه ای است. تقریباً 50 درصد از افراد مبتلا به آمیلوئیدوز ثانویه آرتریت روماتوئید به عنوان بیماری زمینه ای دارند.
آمیلوئیدوز خانوادگی (Familial amyloidosis) به دلیل ناهنجاری در ژن یکی از چندین پروتئین ایجاد میشود. شایع ترین شکل آمیلوئیدوز ارثی توسط یک نوع بیماری زا در ژن ترانس تیرتین (transthyretin یا TTR) ایجاد میشود که منجر به آمیلوئیدوز ATTRv میشود. بیش از 100 نوع مختلف در ژن transthyretin گزارش شده است و رایج ترین نوع آن V30M نامیده شده است.
انواع مختلف ژن TTR با آمیلوئیدوز مرتبط است که بر سیستمهای مختلف اندام تاثیر میگذارد. به ندرت، انواع ژنهای پروتئینهایی که باعث آمیلوئیدوز میشوند عبارتند از فیبرینوژن A زنجیره آلفا (fibrinogen A alpha chain)، آپولیپوپروتئین (apolipoprotein) A1 و A2، ژلسولین (gelsolin)، LECT2 و سیستاتین C (cystatin C).
همه آمیلوئیدوزهای ارثی به دنبال توارث اتوزومال غالب هستند. بیشتر بیماریهای ژنتیکی بر اساس وضعیت دو نسخه از یک ژن، یکی از پدر و دیگری از مادر، تعیین میشود. اختلالات ژنتیکی غالب زمانی رخ میدهد که تنها یک نسخه از یک ژن غیر طبیعی برای ایجاد یک بیماری خاص ضروری باشد. ژن غیر طبیعی میتواند از هر یک از والدین به ارث برسد یا میتواند نتیجه یک جهش جدید (تغییر ژن) در فرد مبتلا باشد. خطر انتقال ژن غیر طبیعی از والدین مبتلا به فرزند برای هر بارداری 50 درصد است. این خطر برای مردان و زنان یکسان است. با این حال، هر فردی که این ژن را دریافت میکند، در نهایت به آمیلوئیدوز مبتلا نمیشود.
علت دقیق آمیلوئیدوز بتا 2 میکروگلوبولین مرتبط با دیالیز به طور کامل شناخته نشده است. یک کلیه با عملکرد طبیعی میتواند بتا 2 میکروگلوبولین را پاک کند. در برخی از افراد تحت دیالیز طولانی مدت یا برخی از افراد تحت دیالیز صفاقی متحرک مداوم (CAPD یا continuous ambulatory peritoneal dialysis)، ناتوانی کلیهها در عملکرد صحیح منجر به احتباس غیر طبیعی و تجمع پروتئین بتا-میکروگلوبولین میشود.
برخی از افراد مبتلا به نارسایی کلیه در مرحله نهایی نیز به این شکل از آمیلوئیدوز مبتلا شده اند. اگرچه اعتقاد بر این است که این احتباس و انباشت عامل اصلی است، عوامل دیگری برای ایجاد این اختلال مورد نیاز است، به همین دلیل است که تنها درصدی از افراد دیالیز به آمیلوئیدوز بتا 2 میکروگلوبین مربوط به دیالیز مبتلا میشوند.
جمعیتهای آسیب دیده
تخمین زده میشود که سالانه حدود 4000 مورد جدید آمیلوئیدوز AL در ایالات متحده وجود دارد، اگرچه بروز واقعی ممکن است در نتیجه تشخیص نادرست و یا تشخیص اشتباه تا حدودی بیشتر باشد. در حالی که تصور میشود میزان بروز در مردان و زنان برابر است، حدود 60 درصد از بیماران مراجعه کننده به مراکز آمیلوئید مرد هستند. آمیلوئیدوز AL در افراد 20 ساله گزارش شده است اما معمولا در سنین 50 تا 65 سالگی تشخیص داده میشود.
افراد در معرض خطر ابتلا به آمیلوئیدوز AA شامل افرادی هستند که بیماریهای التهابی مزمن مانند آرتریت روماتیسمی (rheumatic arthritis)، آرتریت پسوریاتیک (psoriatic arthritis)، آرتریت مزمن نوجوانان (chronic juvenile arthritis)، اسپوندیلیت آنکیلوزان در کودکان (ankylosing spondylitis in children)، بیماری التهابی روده و تب مدیترانه ای خانوادگی (familial Mediterranean fever) دارند. افراد مبتلا به بیماریهای عفونی مزمن مانند سل، جذام، برونشکتازی (bronchiectasis)، استئومیلیت مزمن (chronic osteomyelitis) و پیلونفریت مزمن (chronic pyelonephritis) نیز در معرض خطر هستند. آمیلوئیدوز AA در کمتر از 5 درصد از افراد مبتلا به این شرایط رخ میدهد.
آمیلوئیدوز خانوادگی ناشی از یک نوع ژن ترانس تیرتین تقریباً در 1 نفر از هر 100000 قفقازی در ایالات متحده و بیشتر در آمریکاییهای آفریقایی تبار (تقریباً 4 درصد در آن جمعیت) رخ میدهد. این بیماری در پرتغال، سوئد، ژاپن، ایرلند، اسپانیا، فرانسه، فنلاند، آلمان و یونان شایع است. علائم معمولا بین 40 تا 65 سالگی شروع میشود.
اگرچه آمیلوئیدوز خانوادگی و AA احتمالاً کمتر از آمیلوئیدوز AL شایع هستند، آمیلوئیدوز ATTRwt احتمالاً شایع تر است اما به طور قابل توجهی کمتر تشخیص داده میشود.
اختلالات با علائم مشابه
اختلالات زیر ممکن است با آمیلوئیدوز همراه باشد. آمیلوئیدوز ممکن است همراه با یا در نتیجه اختلالات زیر ظاهر شود:
مولتیپل میلوما (Multiple myeloma)، لنفوم هوچکین (Hodgkin lymphoma)، لنفوم (lymphoma)، کارسینوم مدولاری تیروئید (medullary carcinoma of the thyroid)، بیماری ویپل (Whipple disease)، بیماری کرون (Crohn disease)، استئومیلیت (osteomyelitis)، آرتریت روماتوئید (rheumatoid arthritis)، اسپوندیلیت انکیلوزان (ankylosing spondylitis)، سندرم رایتر (Reiter syndrome)، آرتریت پسوریاتیک (psoriatic arthritis)، سل (tuberculosis)، جذام زودرس (leprosy)، بیماری مایکروگلوبولومیور (familial Mediterranean fever)، بیماری مایکروگلوبولایورم آنمی (Waldenstrom macroglobulinemia).
تشخیص
به ویژه در مورد آمیلوئیدوز AL، تشخیص زود هنگام کلید بقا و بازیابی کیفیت زندگی پس از درمان است. تشخيص آميلوييدوز به دنبال شرح حال دقيق بيمار و ارزيابي باليني مشكوك است اما نيازمند آسپيراسيون پد چربي شكمي (abdominal fat pad) و يا بيوپسي (biopsy) از اندام درگير است.
اگر به دلایل بالینی مشکوک به بیماری باشد، بیوپسی از اندام درگیر بیشترین بازده را خواهد داشت. ماده بیوپسی به صورت میکروسکوپی بررسی میشود و با رنگی به نام قرمز کنگو (Congo red) رنگ میشود که در صورت وجود آمیلوئید در میکروسکوپ پلاریزه کننده رنگ سبز ایجاد میکند. هنگامی که آمیلوئیدوز در بیوپسی بافت تشخیص داده میشود، ضروری است که فرد مبتلا بیشتر مورد ارزیابی قرار گیرد تا مشخص شود چه اندامهایی تحت تأثیر قرار گرفته اند.
هنگامی که بیوپسی بافت آمیلوئیدوز ایجاد شد، تعیین نوع آمیلوئیدوز بسیار مهم است. در آمیلوئیدوز AL، تظاهرات دیسکرازی سلولهای پلاسما در 98 درصد موارد مشاهده میشود. در 2 درصد موارد، لنفوم سلول B به عنوان عامل آمیلوئیدوز AL شناسایی میشود.
آزمایشهای خاصی که برای تشخیص دیسکرازی سلولهای پلاسما یا کلون سلول B استفاده میشوند، عبارت اند از:
- ایمونوفیکساسیون (immunofixation) و الکتروفورز پروتئین (protein electrophoresis) خون و ادرار
- بیوپسی مغز استخوان (bone marrow biopsy) با رنگ آمیزی ایمونوشیمیایی سلولهای پلاسما (immunochemical staining of plasma cells) برای زنجیرههای سبک کاپا و لامبدا و بررسی زنجیره سبک بدون سرم.
سنجش زنجیره سبک تشخیص آمیلوئیدوز ارثی TTR را میتوان با انجام آزمایش ژنتیک مولکولی برای انواع ژن TTR بر روی نمونه خون تایید کرد. در غیاب انواع ترانس تیرتین (transthyretin)، اشکال بسیار نادری از آمیلوئیدوز خانوادگی ممکن است وجود داشته باشد.
اگر بیمار یک مرد مسن با درگیری قلبی ایزوله شده از نظر بالینی باشد، محتمل ترین تشخیص، آمیلوئیدوز ATTRwt است، وضعیتی که در آن ترانس تیرتین طبیعی در قلب رسوب میکند.
رنگ آمیزی ایمنی اختصاصی (به عنوان مثال، میکروسکوپ الکترونی ایمونوگلد یا immunogold electron microscopy) بافت به طور مناسب حفظ شده در مراکز تخصصی موجود است و ویژگی بالایی برای تعیین نوع دقیق آمیلوئید ارائه میدهد. در موارد تشخیصی دشوار، طیف سنجی جرمی (mass spectrometry) قادر است ساختار مولکولی رسوبات آمیلوئید را دقیقاً تعیین کند – این تکنیک بیشتر و بیشتر مورد استفاده قرار میگیرد. روشی به نام اسکن آمیلوئید P سرم نشاندار شده (radiolabeled serum amyloid P یا SAP) در چند مرکز در اروپا که در آمیلوئیدوز تخصص دارند موجود است. این آزمایش برای نظارت و ارزیابی میزان تجمع رسوبات آمیلوئید استفاده میشود.
در افراد تحت دیالیز طولانی مدت یا با نارسایی کلیه در مرحله نهایی، ممکن است تستهای آزمایشگاهی انجام شود که میتواند نمونههای خون یا ادرار را برای تشخیص افزایش سطح پروتئین B2M تجزیه و تحلیل کند.
روشهای درمانی
نوع درمان موجود بر اساس نوع آمیلوئیدوز و وضعیت بالینی بیمار هدایت میشود. در آمیلوئیدوز AL، علت سلولهای پلاسما غیر طبیعی است و به همین دلیل، شیمی درمانی (chemotherapy) با هدف از بین بردن آن سلولها سنگ بنای درمان را تشکیل میدهد. رژیمهای مختلفی مورد مطالعه قرار گرفته اند اما رژیمهایی که بیشترین شواهد تاریخی را دارند، ملفالان (melphalan) و دگزامتازون (dexamethasone) هستند که به صورت خوراکی یا ملفالان با دوز بالا به صورت داخل وریدی با پیوند سلولهای بنیادی اتولوگ (autologous stem cell transplantation) تجویز میشوند. هر دو به یک اندازه موثر هستند اما درمانها و عوارض جانبی متفاوت هستند.
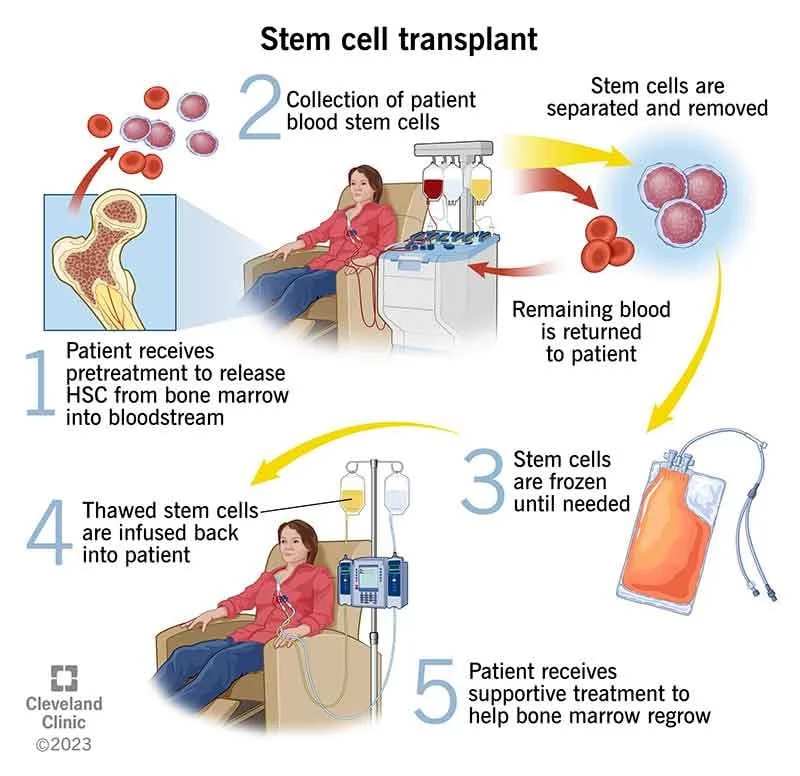
ملفالان با دوز بالا همراه با پیوند سلولهای بنیادی (stem cell transplantation) یک درمان درگیر است که اغلب شامل 2 تا 3 هفته بستری در بیمارستان و چند ماه زمان بهبودی اضافی است. استفاده از ملفالان خوراکی به صورت ماهانه کمتر سمی است اما با خطر بالاتر سرطان خون مرتبط با درمان همراه است. عوامل جدیدتر فعال در مولتیپل میلوما (یکی دیگر از بیماریهای سلولهای پلاسما غیر طبیعی)، مانند Velcade (bortezomib) یا Revlimid (لنالیدومید یا lenalidomide)، همچنین در آمیلوئیدوز AL بسیار مؤثر هستند و نشان داده شده است که در بیماران مبتلا به بیماری عود کننده سودمند هستند.
اغلب، این داروها در درمان اولیه گنجانده میشوند. ترکیب بورتزومیب (bortezomib)، سیتوکسان (سیکلوفسفامید یا cyclophosphamide) و دگزامتازون (dexamethasone) با تحمل خوب و پاسخ سریع همراه است. اخیراً، آژانسهای نظارتی ترکیب CyBorD و Daratumumab را برای درمان آمیلوئیدوز AL تأیید کرده اند و این اکنون استاندارد مراقبت برای بیماران با تشخیص جدید است. درمان خاص برای هر فردی باید با شرایط منحصر به فرد او شخصی سازی شود.
دو عامل مهم تعیین کننده بقای طولانی مدت با AL، وجود و میزان درگیری قلبی و پاسخ خونی به درمان است.
درمان حمایتی (درمان نارسایی احتقانی قلب، توجه به تغذیه، درمان نوروپاتی اتونومیک و غیره) یک اقدام همزمان بسیار مهم است. با توجه به پیچیدگی بیماری، توصیه میشود که درمان در مرکزی با تجربه آمیلوئیدوز انجام شود یا حداقل بیمار باید در چنین مرکزی ارزیابی اولیه را انجام دهد و در طول درمان در جامعه محلی ارتباط مستمر داشته باشد.
آمیلوئیدوز ارثی TTR در صورت امکان با حذف منبع تولید غیر طبیعی TTR درمان میشود. از آن جایی که منبع غالب کبد است، پیوند کبد در بیمارانی که با دقت انتخاب شدهاند، انجام میشود که بیماری آنها خیلی پیشرفت نکرده است. Onpattro (patisiran) و Tegsedi (inoteresen) خاموش کنندههای ژن TTR هستند و توسط سازمان غذا و داروی آمریکا ( یا FDA) برای درمان آمیلوئیدوز ATTRv با نوروپاتی محیطی تایید شده اند. در سال 2019، FDA Vyndaqel (tafamidis meglumine) را برای درمان کاردیومیوپاتی یا cardiomyopathy (بیماری قلبی) ناشی از ATTR (ATTR-CM) تایید کرد.
مشاوره ژنتیک برای افراد مبتلا به آمیلوئیدوز ارثی و اعضای خانواده آنها توصیه میشود.
در آمیلوئیدوز ATTRwt، درمان حمایتی است اما هم برای این بیماری و هم برای ATTR، درمانهای دارویی با هدف تثبیت مولکول ترانس تیرتین و در نتیجه جلوگیری از تشکیل آمیلوئید به طور فعال بررسی میشوند.
پایه اصلی درمان آمیلوئیدوز AA، درمان بیماری زمینه ای است. پیوند کلیه برای بیماری کلیوی ناشی از آمیلوئیدوز AA با موفقیت انجام شده است.
در سال 2015، FDA استفاده از دستگاه پزشکی به نام Lixelle Beta 2-microglobulin ستون آفرزیس را برای درمان آمیلوئیدوز بتا2-میکروگلوبولین مرتبط با دیالیز مجاز کرد. این دستگاه با حذف پروتئین بتا 2m از خون کار میکند.
همچنین بخوانید:
- طیف سنجی جرمی (Mass Spectrometry (MS)): اصول، طرز کار، ابزارها، مراحل و موارد استفاده
- بیماری کرون چیست؟
- مولتیپل میلوما (Multiple Myeloma) چیست؟ انواع، علائم، تشخیص و درمان
- استئوسارکوم (Osteosarcoma) چیست؟ علائم، تشخیص، پیشگیری و درمان
- سارکوم بافت نرم چیست؟ انواع، علائم، تشخیص و درمان
مترجم: فاطمه فریادرس
آمیلوئید در کجا ساخته میشه؟
آمیلوئید در کبد، مغز استخوان و سلولهای التهابی مثل ماکروفاژ ساخته میشود و سپس در بافتهای مختلف بدن رسوب میکند.
ایا در کودکان ده یا دوازده ساله هم آمیلوئیدوز قلبی اتفاق می افتد؟
آمیلوئیدوز قلبی در کودکان ۱۰–۱۲ ساله بسیار نادر است و معمولاً در سنین بالا دیده میشود.
در کودکان، فقط در بیماریهای ارثی خاص یا التهابی مزمن شدید ممکن است رخ دهد.
سلام بابام درگیر این مریضی شده برای درمان باید پیش کدام متخصص مراجعه کنیم
برای درمان آمیلوئیدوز، ابتدا نوع بیماری و اندامهای درگیر باید شناسایی شوند. به طور کلی، شروع با یک متخصص داخلی یا هماتولوژیست میتواند بهترین نقطه شروع باشد، و سپس بر اساس ارزیابیهای بیشتر، به متخصصان دیگر ارجاع داده خواهید شد. مراجعه سریع به پزشک و پیگیری دقیق درمان برای مدیریت این بیماری بسیار مهم است.